|
|
|
Indian Pediatr 2020;57: 239-253 |
 |
Refractory and Super-refractory Status
Epilepticus
|
|
Debopam Samanta1, Lisa Garrity2
and Ravindra Arya2,3
From
1Child Neurology Division, Department of Pediatrics,
University of Arkansas for Medical Sciences, Little Rock, Arkansas;
2Comprehensive Epilepsy Center, Division of
Neurology, Cincinnati Children’s Hospital Medical Center, Cincinnati,
Ohio; and 3Department of Pediatrics,
University of Cincinnati College of Medicine, Cincinnati, Ohio; USA.
Correspondence to: Dr Ravindra Arya,
Division of Neurology, Cincinnati Children’s Hospital Medical
Center, MLC 2015, 3333 Burnet Avenue, Cincinnati, Ohio, 45229
USA.
Email:
[email protected]
|
|
Context: Refractory
status epilepticus (RSE) and super-refractory status epilepticus (SRSE)
are neurological emergencies with considerable mortality and morbidity.
In this paper, we provide an overview of causes, evaluation, treatment,
and consequences of RSE and SRSE, reflecting the lack of high-quality
evidence to inform therapeutic approach. Sources: This
is a narrative review based on personal practice and experience.
Nevertheless, we searched MEDLINE (using PubMed and OvidSP vendors) and
Cochrane central register of controlled trials, using appropriate
keywords to incorporate recent evidence. Results:
Refractory status epilepticus is commonly defined as an acute convulsive
seizure that fails to respond to two or more anti-seizure medications
including at least one non-benzodiazepine drug. Super-refractory status
epilepticus is a status epilepticus that continues for ³24 hours despite
anesthetic treatment, or recurs on an attempted wean of the anesthetic
regimen. Both can occur in patients known to have epilepsy or de novo,
with increasing recognition of autoimmune and genetic causes.
Electroencephalography monitoring is essential to monitor treatment
response in refractory/super-refractory status epilepticus, and to
diagnose non-convulsive status epilepticus. The mainstay of treatment
for these disorders includes anesthetic infusions, primarily midazolam,
ketamine, and pentobarbital. Dietary, immunological, and surgical
treatments are viable in selected patients. Management is challenging
due to multiple acute complications and long-term adverse consequences.
Conclusions: We have provided a synopsis of best
practices for diagnosis and management of refractory/super-refractory
status epilepticus and highlighted the lack of sufficient high-quality
evidence to drive decision making, ending with a brief foray into
avenues for future research.
Keywords:
Convulsive status epilepticus, Epilepsy, Management, Outcome.
|
|
Status epilepticus (SE) is a common pediatric neurological emergency.
The definition of SE has evolved over time, to reconcile the likelihood
of spontaneous seizure cessation based on pathophysiology, versus the
operational urgency to achieve seizure termination and avoid adverse
consequences. We have summarized some of the key definitions in
Box I. The temporal evolution of SE is conventionally divided
into early (5-30 min), established (30-60 min), and refractory (≥60 min) phases [1].
Box I Conceptual Evolution of the Definition of
Status Epilepticus and Other Related Entities
Conceptual basis for definition of status epilepticus
• Mechanistic: failure of homeostatic mechanisms to
control a seizure.
• Physiological: ictal compromise
of neuronal survival.
• Operational: ictal duration
to warrant emergent treatment.
Definition of status
epilepticus
• 1904: Seizures occur so frequently that
coma and exhaustion are continuous between seizures.
(Clark and Prout)
• 1940: Severest form of seizures
in which the post-convulsive sleep of one attack is cut
short by development of the next (seizure). (Wilson)
• 1964: Seizure persists for a sufficient length of time
or is repeated frequently enough to produce a fixed and
enduring epileptic condition. (ILAE 1st International
Classification of Epileptic Seizures)
• 1981: Seizure
persists for a sufficient length of time or is repeated
frequently enough that recovery between attacks does not
occur. (ILAE revision)
• 1993: More than 30 minutes
of (1) continuous seizure activity or (2) two or more
sequential seizures without full recovery of
consciousness between seizures. (AES working group on
SE)
• 1998: Two or more generalized convulsions,
without full recovery of consciousness between seizures,
or continuous convulsive activity for more than 10
minutes (overt generalized convulsive SE). (Veterans
Affairs Study)
• 1999: Operational definition:
generalized convulsive SE in adults and children >5
years old refers to ³5 min of continuous seizures, or ³2
discrete seizures between which there is incomplete
recovery of consciousness Mechanistic definition:
failure of the “normal” factors that serve to terminate
a typical generalized tonic-clonic seizure. (Lowenstein,
Bleck, and Macdonald)
• 2015: A condition resulting
either from the failure of the mechanisms responsible
for seizure termination or from the initiation of
mechanisms which lead to abnormally prolonged seizures
(after time point t1). It is a condition that can have
long-term consequences (after time point t2), including
neuronal death, neuronal injury, and alteration of
neuronal networks, depending on the type and duration of
seizures.Where: for tonic-clonic SE (t1=5 min, t2=30
min), focal SE with impaired consciousness (t1=10 min,
t2>60 min), and absence SE (t1=10-15 min, t2 unknown)
(ILAE Task Force on Classification of SE)
Other
entities
• Refractory SE: SE that fails to respond to
adequately used 1st (BDZ) and 2nd line (appropriately
chosen non-BDZ) medications.
• Super-refractory SE:
SE that persists for 24 hours or more after initiation
of 3rd line medications (anesthetics) or recurrence of
seizure during withdrawal of the anesthetics.
•
NORSE: New onset of refractory SE without a clear acute
or active structural, toxic, or metabolic cause in a
patient without active epilepsy or other preexisting
relevant neurological disorder. It is a clinical
presentation and not a specific diagnosis.
• FIRES:
NORSE with a prior febrile infection between 2 weeks and
24 hours prior to onset of refractory SE, with or
without fever at the actual onset of SE. It is a
sub-category of NORSE.
AES: American Epilepsy
Society; ILAE: International League Against Epilepsy;
FIRES: Febrile Infection Related Encephalopathy
Syndrome; NORSE: New-Onset Refractory Status
Epilepticus; SE: Status Epilepticus]. |
There are two common definitions of refractory SE
(RSE), which may operationally converge. These include a
convulsive seizure lasting longer than 60 minutes, which may be
continuous or intermittent without return to baseline mental
status; and an acute convulsive seizure that fails to respond to
³2 anti-seizure medications (ASMs) including at least one
non-benzodiazepine ASM. In our experience, the latter criteria
is more commonly used in practice. At present, there is a dearth
of sufficient high-quality evidence to formulate a uniform
management strategy for RSE, resulting in variability in
treatment approaches and the choice of therapeutic endpoint(s).
Partly because of this, about 15%-35% of RSE patients fail to
achieve desired treatment response, and progress to
super-refractory SE (SRSE), which is defined as an SE that
continues for 24 hours or more despite anesthetic treatment, or
recurs on an attempted wean of the anesthetic regimen [2,3].
We, herein, provide an overview of RSE and SRSE. However, we
have not reviewed the epidemiology of RSE/SRSE, because of
insufficient data for these entities separate from SE. We have
also not discussed the pathophysiology of drug-resistance in SE,
because the relative importance of different molecular
mechanisms remains uncertain at present, and does not inform
treatment approach. Finally, our discussion is focused on
RSE/SRSE evolving from a convulsive seizure with impaired
awareness, and does not address epilepsia partialis continua.
Causes and Risk Factors
The
underlying causes of RSE/SRSE in a patient with no previous
history of seizures, or possible triggers which may precipitate
RSE/SRSE in a patient known to have epilepsy, are similar to
those responsible for the more common convulsive SE. In general,
any pathology which can trigger an acute symptomatic seizure can
cause an SE which may progress to an RSE/SRSE. This includes
neurological and systemic infections, acute vascular events,
traumatic brain injury, and immune, metabolic, or toxic
encephalopathies.
Febrile SE: Lessons from
FEBSTAT
In the pediatric population,
prolonged febrile seizures constitute the most common subgroup
and account for up to 35% of all episodes of SE [4]. The
importance of recognizing and promptly managing febrile SE has
been underscored by the FEBSTAT study. In this study, 71% of
patients with acute changes on brain magnetic resonance imaging
(MRI) after febrile SE, were found to have obvious hippocampal
sclerosis 1 year later [5,6]. Given the importance of
hippocampal sclerosis as a substrate for drug-resistant temporal
lobe epilepsy, this finding emphasizes the direct association
between the two. Another important finding from FEBSTAT study
was the association of seizure duration with the time to initial
treatment with benzodiazepines (BDZs), where every 2.7 min delay
in the initial treatment was associated with 1.3 min increase in
seizure duration [6,7]. Another study which found shorter median
duration of febrile SE (35 min) compared to FEBSTAT (68 min),
potentially attributed it to shorter time to access emergency
medical services and initial treatment [8].
Infectious Causes
The etiology of RSE/SRSE
varies geographically, and studies from India have noted a
predominance of infectious causes [9-11]. In a series of 148
adults with encephalitis, 18 were diagnosed with SE,
predominantly in those with herpes simplex virus (HSV) infection
or Japanese encephalitis [12]. Children were noted to be more
susceptible to have encephalitis-related SE in this cohort. The
diagnosis of HSV is important in SE/RSE patients, as treatment
with acyclovir within 24 hours of onset has been shown to be
associated with better prognosis [13]. A recent study reported
convulsive SE in 41 patients with neurocysticercosis, although
they did not progress to RSE/SRSE [14]. The duration of SE was
found to be shorter in patients with single calcific lesion
compared to those with degenerating cysts. In another study
including 141 children presenting emergently with acute
convulsive seizures, 49% were found to have neurocysticercosis,
though again the progression to RSE was not reported [11]. There
is a paucity of data on epidemiology of RSE/SRSE from India in
other common infections including acute bacterial
meningoencephalitis, cerebral malaria, and dengue.
Differentiating acute symptomatic SE in a young child with a
neurological infection, from a febrile SE, is a challenging and
important consideration.
Autoimmune and Genetic
Etiologies
Regarding RSE/SRSE, it is important
to recognize two particular groups of diseases, which may prompt
an etiology-specific treatment or prognostication early in the
course of management. The first group includes autoimmune
encephalitis. In children and adolescents, the leading
autoimmune entity is anti-NMDA-receptor encephalitis, which can
present with non-specific manifestations, such as, fever or
headache in the prodromal stage. The next stage, involving the
cerebral cortex, is the one which can present with RSE/SRSE in
additional to behavioral symptoms. In children, these behavioral
symptoms can take the form of new-onset temper tantrums in an
otherwise well-adjusted child, unexplained episodes of
aggression, and speech disturbances. Compared to the predominant
temporal localization for seizures in adults with anti-NMDAR
encephalitis, extra-temporal seizures, and sometimes even
diffuse bilateral ictal onset can be seen in children, and
should not exclude this consideration [15,16]. The other limbic
encephalitis are also being increasingly recognized in children,
and may often not occur as a para-neoplastic syndrome. In
addition to seizures, which can progress to SE/RSE, other
manifestations of limbic encephalitis can be relatively
non-specific in children. The behavioral phenotype of limbic
encephalitis in adults often consists of disturbances in recent
memory, and affective symptoms including irritability,
depression, or hallucinations. However, in children,
non-specific disturbances of executive function (attention
control, social inhibition, regulation of purposeful behavior)
are often seen, and should be evaluated in a developmental
context [17]. The second important etiologic subgroup of
RSE/SRSE, which should be evaluated promptly, comprises of
certain genetic epilepsies. These include, but are not limited
to, Dravet syndrome and other sodium channelopathies, ring
chromosome 20, pathogenic variants in polymerase-G or
amino-acyl-tRNA synthetase genes affecting mito-chondrial
function, and Angelman syndrome [18].
Seizure
Triggers
About 16-38% SE episodes occur in
children with a prior diagnosis of epilepsy, with low
anti-seizure medication levels being the most common risk factor
[4]. Low levels of regular anti-seizure medicines can, in turn,
result from lack of adherence, scheduled withdrawal,
insufficient dose, interaction with other concurrent
medications, or growth spurt, etc. Other common precipitants of
SE include inter-current illness, exposure to a known trigger
(e.g. sleep deprivation in some idiopathic generalized epilepsy
syndromes), or metabolic decompensation.
Diagnostic Evaluation
The goals of evaluation
in RSE/SRSE include diagnostic verification, particularly in
patients with subtle and non-convulsive SE (NCSE); monitoring
for therapeutic efficacy; diagnosis of the underlying etiology
and/or risk factors; and early recognition of multisystem
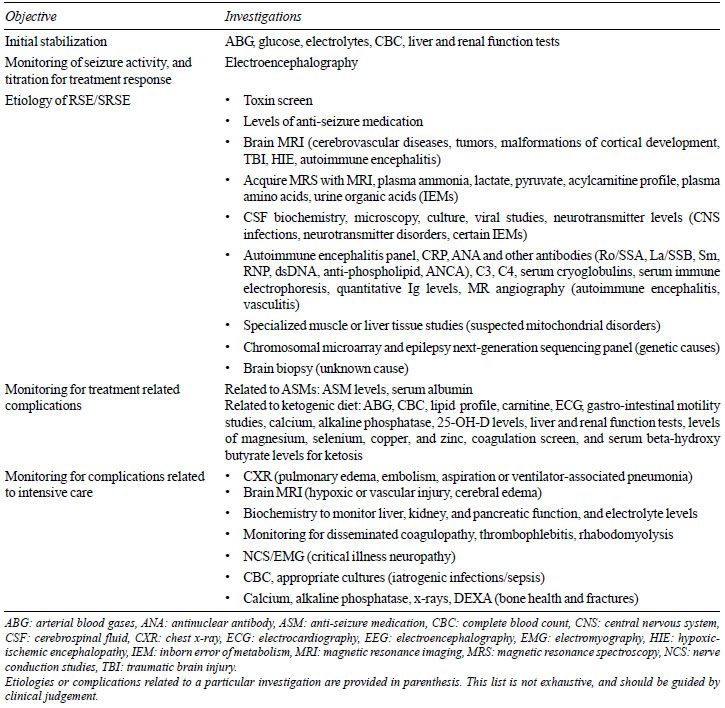
complications. We have summarized these in Table I,
and briefly discuss the role of electroencephalography (EEG) and
neuroimaging below.
Table I Selected
Investigations Useful in Management of Refractory and
Super-refractory Status Epilepticus
|
 |
EEG
Monitoring
The foremost utility of EEG
monitoring is to detect ongoing subclinical seizure or NCSE
after the convulsive seizure has apparently terminated. Hence,
professional societies have recommended continuous EEG
monitoring to be initiated within 1 hour of SE, and continued
for at least 24-48 hours, often longer in the presence of
altered consciousness [19-21]. Although the EEG diagnosis of
NCSE has been fraught with disagreement, recent studies have
shown good diagnostic validity and inter-rater agreement for the
Salzburg criteria [22,23]. In patients without known epileptic
encephalopathy, occurrence of >2.5 Hz epileptiform dischargesmay
be sufficient for the diagnosis of NCSE. In case of epileptiform
discharges £2.5 Hz, or the ictal pattern limited to rhythmic
slow waves, it is important to evaluate for
electroencephalo-graphic improvement with a bolus of intravenous
BDZ; associated subtle clinical manifestations; and typical
spatiotemporal evolution in the morphology, frequency, or locus
of the electroencephalographic pattern. The availability of
synchronized video monitoring, allowing simultaneous review of
video-EEG is important in such situations. The diagnosis of NCSE
becomes more challenging in patients with a known epileptic
encephalopathy. In such patients, diagnosis of NCSE requires
evidence of a distinct increase in the locus, field, or
frequency of epileptiform discharges, and an obvious change in
clinical state, or response to an intravenous BDZ bolus [23].
Hence, in these patients, it is important to have a good
understanding of the baseline EEG pattern prior to the onset of
RSE, based on a personal review of previous EEGs.
Other
periodic and rhythmic EEG patterns, which do not meet the
criteria for NCSE, are often labelled as lying on an
interictal-ictal continuum. Although, this terminology is an
useful EEG descriptor, its clinical significance remains
uncertain. However, recognition of periodic patterns including
lateralized periodic discharges (LPDs) or generalized periodic
discharges (GPDs) may be important because of some association
with the risk of seizure recurrence and certain specific
conditions, such as, LPDs with stroke and herpes encephalitis
[24]. Rarely, other EEG patterns may suggest a specific
etiology, for example, extreme delta brushes in anti-NMDAR
encephalitis.
Use of quantitative EEG has been
increasing recently in neuro-critical care practice, due to
integration of analysis software into EEG reading suites [25].
During management of RSE/SRSE, quantitative analysis of EEG
trends may be helpful in evaluation of the suppression ratios
over the course of treatment, recognition of periodic patterns
or seizure recurrence, and comparison of power ratios in
canonical EEG frequency bands. However, there is a paucity of
studies validating use of quantitative EEG for prediction of
outcomes in RSE/SRSE. Hence, it is important to verify any
observations on quantitative trends, with a careful review of
raw EEG. In our practice, the two most common uses of
quantitative EEG in the management of RSE/SRSE include temporal
analysis of suppression ratios, and quantitation of seizure
burden once the specific imprint of a given seizure type is
recognized on the density spectral array or amplitude
integration. A detailed discussion of the methods, utility, and
pitfalls of quantitative EEG is outside the scope of this paper.
The second most important role of EEG monitoring in the
management of RSE/SRSE is to establish the therapeutic target.
In these children, clinical evaluation for continued seizure
activity is often non-informative, and becomes more so as the
anesthetic infusions are increased, or neuromuscular blockers
are used to facilitate airway control. At this point, the EEG
usually shows electrographic bursts interrupted by periods of
relative diffuse amplitude suppression. The optimal EEG target
for treatment of RSE/SRSE is not strictly defined, and have
included, suppression of seizures, varying degrees of
suppression-burst pattern, and nearly complete EEG suppression
[26]. Our practice is to aim for complete suppression of
seizures, and achieve 50%-70% suppression ratio. Once this
therapeutic goal is achieved, EEG monitoring is continued for
24-48 hours, when attempts are first made to wean off the
infusions. EEG monitoring is important at this point to
recognize the occurrence of emergence seizures or bursts with
epileptiform elements, though these are not always treated
individually.
Neuroimaging
The
primary goal of neuroimaging in RSE/SRSE is to help identify the
etiology, which in some cases, may lead to a specific urgent
intervention. Secondly, identification of a potentially
epileptogenic lesion may prompt conside-ration of emergency
epilepsy surgery for RSE/SRSE in centers where such expertise is
available. However, the interpretation of MRI studies early in
the course of an RSE/SRSE can be challenging due to
seizure-related or treatment-related changes. Specifically, it
may be worthwhile to distinguish vasogenic edema reflected by
T2/FLAIR hyper-intensities, from cytotoxic edema predominantly
manifested as diffusion restriction. This is because vasogenic
edema can sometimes point to an underlying etiology, whereas
cytotoxic edema often represents compromised neuronal integrity
from RSE/SRSE. In many patients there is an overlap between the
two, and it is helpful to follow their neuroimaging over time.
It is also important to look for confounding factors, for
example, vigabatrin can be associated with both T2
hyper-intensities and restricted diffusion, predominantly in
corpus callosum and deep gray structures [27,28]. In the FEBSTAT
study, about 10% of children with a single febrile SE episode
were found to have altered signal in the hippocampal region.
After one year, 86% of them showed hippocampal volume loss and
71% had evidence of hippocampal sclerosis [5], emphasizing the
need for longitudinal neuroimaging to ascertain neuronal injury
in patients with RSE/SRSE.
Treatment
The primary goal of treatment is the termination of
clinical and electrographic SE. However, the objectives of
treatment in RSE/SRSE are somewhat different from those in
early/established SE. In patients with early/established SE, the
treatment efforts are focused on rapid seizure control to avoid
neuronal injury. However, by definition, RSE represents a
situation where pathophysiological mechanisms that support
drug-response in established SE have been overwhelmed or
altered, and hence, conventional treatment approaches are
unlikely to be successful. Further, in SRSE, it is very likely
that excitotoxic and other processes which compromise neuronal
survival, have already been activated. Hence, the goal of
treatment in RSE/SRSE is to limit or reverse these processes,
prevent their downstream consequences, and salvage end-organ
function to the maximum possible extent [29]. Clinically, this
means that the treatment plan should encompass not only seizure
control, but to also avoid, anticipate, and manage multisystem
dysfunction resulting from ongoing seizure activity, from
medications, and from prolonged unconsciousness and immobility
[30]. While the intensive care related to RSE/SRSE is outside
the scope of this paper, we have briefly summarized the first
and second line treatment for SE, and then discussed specific
therapeutic approaches to RSE/SRSE in more detail.
First- and Second- line Drug Treatments
The American Epilepsy Society guidelines published in 2016,
have synthesized available evidence for drug-treatment of early
and established SE [31]. There is consensus that BDZs represent
the initial treatment of choice for SE, with equipoise among
intramuscular midazolam (MDZ), intravenous lorazepam (LZP), and
intravenous diazepam (DZP). This equipoise is based on three
class I randomized controlled trials, which are summarized
below. The Veterans Affairs study compared four interventions
including DZP followed by phenytoin (PHT), LZP, phenobarbital
(PHB), and PHT alone [32]. There were no significant differences
among these 4 interventions on intention-to-treat (ITT)
analysis. However, on pairwise comparisons in patients with
generalized convulsive SE, LZP was superior to PHT, for
cessation of all motor and EEG seizure activity within 20 min of
starting drug infusion and no recurrence of seizure activity
during the next 40 min. The Rapid Anticonvulsant Medication
Prior to Arrival Trial (RAMPART), showed equivalence between
intramuscular MDZ and intravenous LZP for the proportion of
patients who achieved seizure termination without need for
additional rescue [33]. However, the auto-injectors used in
RAMPART are not available commercially, and whether conventional
intramuscular MDZ injection will have similar efficacy is open
to question. The third class I trial, showed equivalence between
intravenous LZP and DZP for the proportions of patients with
seizure termination, need for assisted ventilation, recurrence
within 1 hour, and time to SE termination [34].
Only
when these three options are not available, or not feasible due
to challenges in achieving intravenous access in a convulsing
child, other treatments may be considered, including rectal DZP,
intranasal MDZ, and buccal MDZ. A network meta-analysis of 16
studies, specifically looking at non-venous treatments for acute
convulsive seizures, found intramuscular and intranasal MDZ to
be superior to other comparators for clinically relevant
outcomes [35]. Specifically, this meta-analysis excluded data
from RAMPART in their quantitative synthesis, thus independently
supporting the efficacy of conventional intramuscular MDZ and
encouraging the use of intranasal MDZ. From a practical
standpoint, MDZ has the advantage of not needing refrigeration
for storage. In US, MDZ nasal spray has been recently approved
to treat seizure clusters and acute repetitive seizures [36].
Intranasal formulations of DZP and rapid systemic delivery of
alprazolam (staccato alprazolam) by inhalation of thermally
generated aerosol from a single-use hand-held device, are
currently undergoing evaluation [37,38].
Appropriate
first-line drug-treatment results in seizure termination in
about 65% of patients. However, the subsequent choice of
treatment is not driven by rigorous evidence. Traditionally,
intravenous PHT has been the most commonly used 2nd line
treatment for SE. Fosphenytoin (FOS) has been preferred over
PHT, due to the ability for faster infusion, less risk of
cardiac arrhythmias, and decreased incidence of local tissue
reaction. However, a meta-analysis of 22 studies has found the
efficacy of PHT to be below, though not significantly, to that
of valproate (VPA), PHB, and levetiracetam (LEV) [39]. This
meta-analysis also concluded that there is insufficient data for
the use of lacosamide (LAC) in the treatment of SE. Recently,
two different open-label trials: ConSEPT study from Australia
and New Zealand, and EcLiPSE study from United Kingdom, have
compared LEV and PHT in children with BDZ-refractory SE [40,41].
Clinical cessation of seizure activity 5 min after infusions
were not statistically different between the two groups.
Overall, 50%-70% of participants achieved primary efficacy
endpoint. An ongoing trial in US, Established Status Epilepticus
Treatment Trial (ESETT), is also comparing LEV, FOS, and VPA
[42]. Given the lack of class I trials, decisions about 2nd line
treatment are often based on local availability, cost, and most
importantly patient-specific factors.
In addition to
medical stabilization, the treatment of early/established SE
proceeds concurrently with attempts to ascertain the etiology of
SE. The specific treatments are driven by presumptive or
definite etiology, often determined by local epidemiology, and
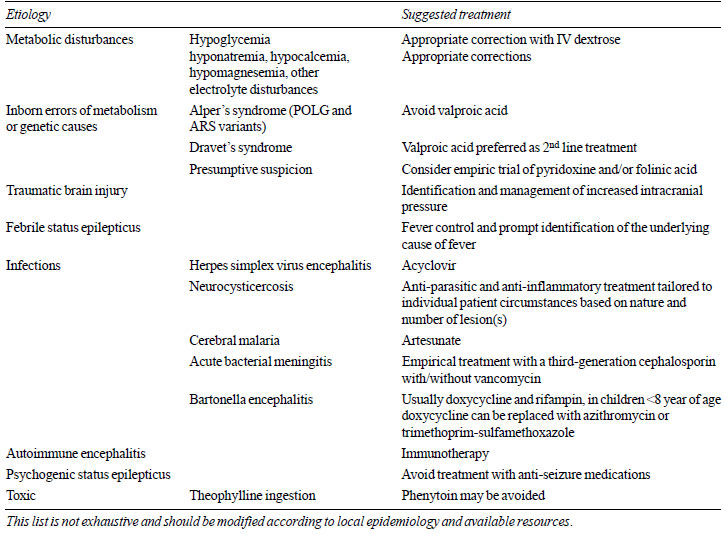
investigative resources. A few suggestions are offered in
Table II.
| Table II Suggested
Etiology-Specific Treatments in Status Epilepticus |
 |
Third-line Treatment:
Drug Therapy for RSE
There is a further paucity
of clinical trials for drug treatment of RSE. Designing
comparative effectiveness trials in this population has been
challenging due to several reasons, including heterogeneity of
1st and 2nd line treatment choices, concurrent use of multiple
other medications affecting the nervous system, variability in
the time course of administration of different medications, and
the level of supportive intensive care. This is exemplified by a
multicenter trial which was terminated inconclusively, because
only 14 patients could be recruited over 3 years, against an
estimated sample size of 150 [43]. Another trial of brexanolone
(an aqueous formulation of the neuroactive steroid
allopregnanolone) in 25 patients with SRSE was also inconclusive
regarding efficacy end point, though it showed reasonable
tolerability [44].
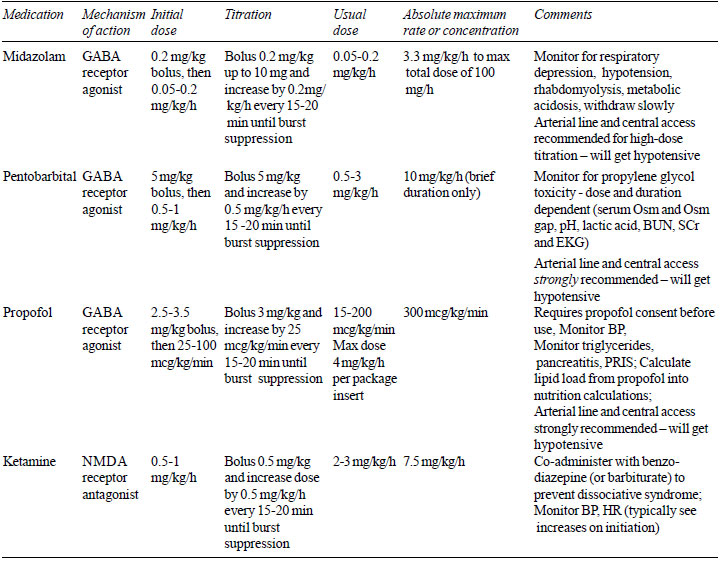
Conventionally, the three most
commonly used anesthetic agents for the treatment of RSE include
MDZ, short-acting barbiturates (pentobarbital/thiopentone), and
propofol (Table III). Currently, MDZ is perhaps
the most commonly used drug for RSE due to faster onset of
action and short duration of effect [45]. However, use of MDZ in
RSE/SRSE is fraught with several issues including development of
tolerance, prolonged half-life with continued use, and potential
for interactions with other drugs, nephrotoxicity,
hepatotoxicity, and cardio-respiratory depression [45,46].
Several studies have reported on the use of MDZ for RSE/SRSE,
using different doses and treatment targets, as reviewed
elsewhere [45]. A meta-analysis including 111 children showed
that MDZ was as effective as other coma inducing medications,
but had a lower mortality [47]. However, another study that
compared MDZ and DZP in 40 children and found a similar efficacy
(86% and 89%, respectively), reported MDZ to be associated with
a higher recurrence (57% vs 16%) and higher mortality (38% vs
10.5%) [48]. Seizure control has been reported as occurring
within 0.3-1.1 hours [45]. Breakthrough seizures have been
reported in 47-57% of patients [49].
| Table III
Continuous Infusions for Treatment of Refractory Status
Epilepticus |
 |
To compare,
barbiturates have been used for the longest period, and believed
to have higher efficacy. A meta-analysis including 193 adults
with RSE compared pentobarbital, MDZ, and propofol. Although
pentobarbital was associated with a significantly lower
incidence of short-term treatment failure, breakthrough
seizures, and the need to change to a different medication, it
was also associated with a significantly higher frequency of
hypotension [50]. One of the important confounding factors in
this study was that the target of treatment in patients
receiving pentobarbital tended to be burst-suppression, while it
was limited to seizure suppression in those treated with MDZ and
propofol. In pediatric RSE/SRSE the efficacy of pentobarbital
has been reported to be 74-100% in heterogeneous studies
[47,51]. Some experts believe that barbiturates have a
neuroprotective effect and have an additional anti-seizure
efficacy from their ability to lower core body temperature [52].
However, barbiturates have more potent cardiorespiratory
suppression, immunosuppression, longer half-life with storage in
the lipid compartment resulting in slow recovery, and problems
of auto-induction and zero-order kinetics. Intravenous
formulations of pentobarbital also contain propylene glycol,
which at the high cumulative doses necessary to achieve burst
suppression, may cause hyperosmolality and lactic acidosis that
can progress to cardiac arrhythmias, refractory hypotension,
renal dysfunction, and multi-organ failure [53]. It is possible
that advances in intensive care, and the increasing practice for
pre-emptive control of ventilation and perfusion, may help
mitigate some of these challenges.
In adults, propofol
is used for management of RSE/SRSE due to its quick onset of
action and prompt recovery on withdrawal. Studies in adults have
shown that propofol infusion terminates RSE/SRSE in 67% of
patients [54]. Although propofol induces burst-suppression
within 35 minutes of initiation, frequent titration may be
needed to maintain adequate suppression [55]. A retrospective
study of 33 children aged 4 months to 15 years with RSE reported
that propofol was more effective than thiopental in terminating
seizures (64% vs 55%). However, adverse effects including
rhabdomyolysis and hypertrigly-ceridemia, prompted
discontinuation in 18% of patients, with recovery after
discontinuation of propofol [56]. Use of propofol has been
restricted in children due to the risk of propofol infusion
syndrome (cardiac failure, rhabdo-myolysis, metabolic acidosis,
renal failure, and sometimes death) and propensity to induce
dyskinesias which can mimic breakthrough seizures [57]. Risk
factors reported to be associated with a higher likelihood of
the infusion syndrome include high doses, prolonged use,
concurrent use of catecholamines and corticosteroids, and
possibly malnutrition. However, there have been some concerns
that this syndrome may not be limited to propofol, and could in
fact, result from drug-induced cerebral suppression [58]. Also
relevant to pediatric practice are the reported fatalities on
concurrent use of propofol and ketogenic diet (KD) [59].
Ketamine
Acceptance for the use of
ketamine in RSE/SRSE has been steadily increasing due to
favorable hemodynamics and a different mechanism of action than
conventional anesthetics. Given the GABA receptor endocytosis in
RSE/SRSE, and the NMDA glutamate receptor antagonism of
ketamine, there is a theoretical advantage for its use instead
of, or more commonly in addition to, other anesthetic agents. A
systematic review of 25 class IV studies analyzed 244 SE
episodes (37 in children) treated with ketamine [60]. Although
authors reported 73% of children to ‘respond’, the heterogeneity
in the definition of response was not addressed. Our practice is
to start with MDZ infusion, and optimize it to achieve seizure
suppression and >50% burst suppression. In non-responders, we
then consider ketamine concurrent to MDZ. In patients who do not
respond, we finally start pentobarbital and wean off MDZ and
ketamine (Fig. 1). This is different from published multicenter
experience from US, where ketamine was typically used after
pentobarbital [61]. Salient pharmacological aspects of these
drugs are summarized in Table III.
Therapeutic Target for RSE
As we have
already alluded, there is a lack of consensus regarding optimal
target for treatment of RSE/SRSE. Different therapeutic
endpoints have included seizure suppression, burst suppression,
or electro-cerebral silence. Moreover, the extent of
burst-suppression that is associated with best outcomes, remains
undefined [62]. This results in variability in clinical practice
as shown by a study including 35 adult RSE patients, which
reported that patients remained within the target suppression
range (defined as 65-95%) only 0-29% (median 8%) of the total
time under treatment [63]. Our practice is to increase infusions
rapidly to achieve a suppression ratio of 50-70% and complete
seizure control, as soon as possible after failure of 2nd line
treatment, and to frequently review EEG to ensure that this
degree of suppression is maintained. We try to wean off
anesthetic infusions once this treatment target is maintained
for 24-48 hours. However, this duration is not based on rigorous
evidence, and a shorter duration of burst suppression may be
beneficial in some patients, whereas several cycles may be
needed in patients with SRSE. Longer pharmacological coma of up
to 5-7 days may have to be tolerated in particularly challenging
cases. Simultaneously, additional ASMs preferably with
complimentary mechanisms of action, short half-life, and low
incidence of drug inter-actions, should be added to facilitate
ongoing seizure suppression and smooth weaning of infusions.
Topira-mate, LEV, LAC, and more recently, brivaracetam and
perampanel have been used for this purpose [26].
The
effectiveness of this empirical practice was recently assessed
in a cohort of 111 RSE patients recruited prospectively over two
years in an observational study. MDZ was the most frequently
used initial anesthetic agent (78%), and pentobarbital was most
frequently used agent after MDZ failure (82%) [64]. Treating
physicians used up to four cycles of serial anesthetic therapy
in these patients, and seizure termination was achieved in 94%
patients by the second cycle. However, other studies have shown
regional differences in the use of therapeutic coma for RSE
treatment, lack of effect on overall mortality, and increased
length of hospital stay, related costs, and adverse effects
[65,66].
 |
| Fig.1 A suggested
protocol for management of refractory/super-refractory
status epilepticus. |
A suggested protocol for management of
RSE/SRSE, based on the practice at authors’ institutions is
provided as Fig. 1. This protocol is applicable only to patients
>29 days of age. Given the lack of class I evidence,
modifications to this protocol driven by local epidemiology and
resources are strongly encouraged. Treatments other than
anesthetic infusions may be considered earlier based on
suspected/proven etiology of status epilepticus.
Additional Treatment Options
Patients
with RSE, particularly after failure of first anesthetic
infusion, or those with SRSE, represent a desperate situation
for the clinical team. Hence, myriad approaches are brought upon
to control the RSE/SRSE, often based on limited experience.
These diverse treatments have been discussed elsewhere [67]. The
relative position of these modalities in the treatment of SRSE
has to be individualized according to the patient and the
clinical resources.
Ketogenic Diet
Among these approaches, the most promising appears to be
ketogenic diet (KD), partly due to its potential for undermining
pathophysiology of RSE/SRSE [68,69]. A study including 14
pediatric patients reported electrographic seizure resolution
and ³50% suppression in 10 patients within 7 days of starting KD
[70]. In 11/14 patients, continuous infusions could be weaned
off within two weeks of starting KD. However, the authors noted
under-utilization of and delay in starting KD. Other series with
8-17 patients have also reported seizure resolution within 7
days of starting KD in 20-90% patients [67,70]. KD is typically
administered via enteral route, though ketogenic parenteral
nutrition has been used in some cases, both in 4:1 ratio.
However, ketosis may sometimes be difficult to achieve with
concomitant barbiturate infusions which contain propylene glycol
that is metabolized to lactaldehyde and then to lactic acid.
Common reported adverse effects of KD have included metabolic
derangements like hypoglycemia; and gastrointestinal symptoms
like emesis.
Surgery
In patients
with a potentially epileptogenic brain lesion and concordant
neurophysiology, an urgent epilepsy surgery may be considered,
if such expertise is readily available. However, such decisions
also incorporate the location, size, and nature of MRI
lesion(s); and functional significance of surrounding cortex,
which may require intracranial EEG evaluation and electrical
stimulation mapping. Surgical decision-making becomes more
challenging in MRI-negative RSE/SRSE or discordant
neurophysiological data. Functional imaging is often confounded
in such cases by ongoing ictal activity and concurrent
anesthetic infusions. In some cases, palliative options like
vagus nerve stimulation, or corpus callosotomy may be
considered. Non-invasive neuro-stimulation, particularly with
transcranial magnetic stimulation, are emerging modalities for
interrupting RSE/SRSE, which may be useful in future.
Other Approaches
Among other modalities,
therapeutic hypothermia is the only intervention tested in a
randomized controlled trial for RSE/SRSE management. However,
such trials, including the HYBERNATUS study, found the efficacy
of hypothermia to be no better than placebo for RSE/SRSE, and
raised concerns about its safety [67,71]. Immunotherapy
(including any combination of steroids, intravenous
immunoglobulins, or plasma exchange) may be helpful in known
autoimmune epilepsies or entities with presumed immunological
basis, such as febrile-infection related epilepsy syndrome
(FIRES). At present, there is insufficient evidence for or
against the use of immunotherapy in other RSE/SRSE patients.
Similarly, there is no evidence for use of magnesium and
pyridoxine outside of specific indications. These specific
scenarios include using magnesium for acute convulsive seizures
in eclampsia, and pyridoxine for suspected functional antiquitin
deficiency or isoniazid toxicity.
Inhalational
Anesthetics
There is limited experience for the
use of inhaled anesthetics, primarily isoflurane and dexflurane,
late in SRSE unresponsive to MDZ, propofol, and pentobarbital.
In published cases, both isoflurane and dexflurane were
effective in stopping seizures and achieving burst suppression;
however, seizures recurred frequently after discontinuation.
Complications of isoflurane and dexflurane have included
hypotension, atelectasis, infections, paralytic ileus, and deep
vein thrombosis; with death in 3/7 patients in one series [72].
In a recent case report, two patients with prolonged isoflurane
use showed MRI changes in the thalamus and cerebellum, raising
concerns for neurotoxicity [73]. More frequent hippocampal
changes on MRI were seen in patients receiving isoflurane for
RSE compared to matched controls that received only intravenous
anesthetics; with these changes being related to longer duration
of isoflurane use [74]. Given the logistical difficulties and
high incidence of complications with prolonged use of
inhalational anesthetics, the risk/benefit should be carefully
considered before pursuing this course of therapy.
Consequences of RSE/SRSE
The adverse
consequences of RSE/SRSE can be divided into immediate
complications and longer term neuro-morbidity and mortality (Web
Table I). The interim complications mainly result from
aberrant pathophysio-logical processes that are often initiated
in an attempt to control the prolonged seizure but may fail or
get out-of-hand and result in neuronal injury. Additionally,
patients with RSE/SRSE also face complications resulting from
its treatment and those from prolonged intensive care, as
summarized in Web Table 1. More protracted
consequences of RSE/SRSE are conventionally classified into
short-term (during hospitalization or within 30 days of onset of
SE) and long-term (within 10 years following initial survival 30
days after SE onset).
In a multicenter Canadian study of
374 patients with newly diagnosed epilepsy, occurrence of
convulsive SE in 22 children was associated with poor
health-related quality of life after 24 months follow-up [75].
In general, RSE/SRSE have significantly higher morbidity and
mortality compared to SE of shorter duration. In children, RSE
is associated with mortality in 11%-44% of patients and
long-term neurological deficits in 25%-100% of the survivors
[76-79]. The fatality of convulsive SE was 11% in the north
London study, with cumulative incidence of epilepsy,
intellectual disability, and motor impairment of 25%, 12%, and
5%, respectively in survivors after a median follow-up of 9
years [78,79]. This study also reported motor and intellectual
disability to be more prevalent in patients with pre-existing
epilepsy and neurologic disability. Other relatively smaller
studies of pediatric RSE have reported mortality from 16% to
44%, and sequelae in 25%-100% of survivors [47,76,80].
Neurological sequelae of RSE/SRSE appear to have relatively
higher incidence in infants. In a large prospective study with
mean follow-up of 13.2 months, the incidence of neurological
deficits attributable to convulsive SE was 29% in infants £1
year of age, 11% in children 1-3 years of age, and 6% in
children >3 years of age [81].
The risk of subsequent
development of epilepsy is 25%-40% within 2 years after an
RSE/SRSE episode, with a higher propensity in those with
symptomatic causes [82]. The risk of recurrent SE is estimated
to be about 20% within 4 years of the first RSE episode, with
the highest risk in the first year. Progressive and remote
symptomatic etiologies are associated with a higher risk for
recurrent SE compared to febrile SE or acute symptomatic SE
[83]. Long term adverse behavioral outcomes are also prevalent.
After a mean 8 years of follow-up in the North London cohort,
37% SE patients were noted to have behavioral issues and 28%
received a psychiatric diagnosis such as autism, attention
deficit hyperactivity disorder, pervasive developmental disorder
not otherwise specified, and developmental coordination disorder
[84].
Several studies have reported age and etiology to
be major determinants of both short-term and long-term mortality
in SE [4,85]. A systematic review found mortality to be lower in
children as compared to adults and elderly, with short-term
mortality up to 9% and long-term mortality up to 7% [4]. Amongst
children, infants £1 year of age had the highest short-term
mortality, up to 18% [4]. Most SE-related deaths in the short
term have been noted to occur in children with acute symptomatic
etiology [85]. In a cohort study from Kenya, 23% of children
with confirmed CSE died before discharge, with 75% of the deaths
occurring within 48 hours of onset of SE [86].
Avenues for Future Research
RSE/SRSE presents a
host of important unanswered clinical conundrums which have
significant potential to impact patient care. Perhaps the
foremost among these is to generate randomized, blinded, and
adequately controlled evidence for various treatment regimens.
Secondly, the optimal target for treatment needs to be defined.
This will require a careful evaluation of EEG biomarkers for
outcomes of RSE/SRSE. Thirdly, it is desirable to develop
multicenter registries of RSE/SRSE with longitudinal data, which
can be harnessed for predictive modelling of outcomes. There are
unique challenges and opportunities in this regard for
pediatricians and neurologists in India. There is a need to
generate data about epidemiology, causes, diagnostic, and
therapeutic modalities and their yield in India to develop
targeted strategies for intervention that are specific to local
circumstances.
References
1. Sanchez Fernandez I, Abend NS, Agadi S, An S, Arya R,
Carpenter JL, et al. Gaps and opportunities in refractory status
epilepticus research in children: A multi-center approach by the
Pediatric Status Epilepticus Research Group (pSERG). Seizure.
2014;23:87-97.
2. Shorvon S, Ferlisi M. The treatment of
super-refractory status epilepticus: A critical review of
available therapies and a clinical treatment protocol. Brain.
2011;134:2802-18.
3. Mayer SA, Claassen J, Lokin J,
Mendelsohn F, Dennis LJ, Fitzsimmons BF. Refractory status
epilepticus: frequency, risk factors, and impact on outcome.
Arch Neurol. 2002;59: 205-10.
4. Chin RF, Neville BG,
Scott RC. A systematic review of the epidemiology of status
epilepticus. Eur J Neurol. 2004;11:800-10.
5. Shinnar S,
Bello JA, Chan S, Hesdorffer DC, Lewis DV, Macfall J, et al. MRI
abnormalities following febrile status epilepticus in children:
the FEBSTAT study. Neurology. 2012;79: 871-7.
6. Lewis
DV, Shinnar S, Hesdorffer DC, Bagiella E, Bello JA, Chan S, et
al. Hippocampal sclerosis after febrile status epilepticus: the
FEBSTAT study. Ann Neurol. 2014;75:178-85.
7. Hesdorffer
DC, Shinnar S, Lewis DV, Nordli DR, Jr., Pellock JM, Moshe SL,
et al. Risk factors for febrile status epilepticus: a
case-control study. J Pediatr. 2013;163:1147-51.
8.
Bassan H, Barzilay M, Shinnar S, Shorer Z, Matoth I, Gross-Tsur
V. Prolonged febrile seizures, clinical characteristics, and
acute management. Epilepsia. 2013;54:1092-8.
9. Gulati S,
Kalra V, Sridhar MR. Status epilepticus in Indian children in a
tertiary care center. Indian J Pediatr. 2005;72:105-8.
10. Sasidaran K, Singhi S, Singhi P. Management of acute seizure
and status epilepticus in pediatric emergency. Indian J Pediatr.
2012;79:510-7.
11. Arya R, Gulati S, Kabra M, Sahu JK,
Kalra V. Intranasal versus intravenous lorazepam for control of
acute seizures in children: A randomized open-label study.
Epilepsia. 2011;52:788-93.
12. Misra UK, Kalita J.
Seizures in encephalitis: predictors and outcome. Seizure.
2009;18:583-7.
13. Whitley RJ, Alford CA, Hirsch MS,
Schooley RT, Luby JP, Aoki FY, et al. Vidarabine versus
acyclovir therapy in herpes simplex encephalitis. N Engl J Med.
1986;314:144-9.
14. Murthy JMK, Deshmukh DS. Convulsive
status epilepticus due to different evolutionary stages of
neurocysticercosis - solitary cyticercus granuloma, low cyst
load, and single calcific lesion in an endemic country: Clinical
profile. Seizure. 2019;71:229-32.
15. Graus F, Titulaer
MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical
approach to diagnosis of auto-immune encephalitis. Lancet
Neurol. 2016;15:391-404.
16. Armangue T, Petit-Pedrol M,
Dalmau J. Autoimmune encephalitis in children. J Child Neurol.
2012;27:1460-9.
17. Machado S, Pinto AN, Irani SR. What
should you know about limbic encephalitis? Arq Neuropsiquiatr.
2012;70:817-22.
18. Bhatnagar M, Shorvon S. Genetic
mutations associated with status epilepticus. Epilepsy Behav.
2015;49:104-10.
19. Brophy GM, Bell R, Claassen J,
Alldredge B, Bleck TP, Glauser T, et al. Guidelines for the
evaluation and management of status epilepticus. Neurocrit Care.
2012;17:3-23.
20. Herman ST, Abend NS, Bleck TP, Chapman
KE, Drislane FW, Emerson RG, et al. Consensus statement on
continuous EEG in critically ill adults and children, part I:
Indications. J Clin Neurophysiol. 2015;32:87-95.
21.
Herman ST, Abend NS, Bleck TP, Chapman KE, Drislane FW, Emerson
RG, et al. Consensus statement on continuous EEG in critically
ill adults and children, part II: Personnel, technical
specifications, and clinical practice. J Clin Neurophysiol.
2015;32:96-108.
22. Leitinger M, Trinka E, Gardella E,
Rohracher A, Kalss G, Qerama E, et al. Diagnostic accuracy of
the Salzburg EEG criteria for non-convulsive status epilepticus:
A retrospective study. Lancet Neurol. 2016;15:1054-62.
23. Beniczky S, Hirsch LJ, Kaplan PW, Pressler R, Bauer G,
Aurlien H, et al. Unified EEG terminology and criteria for
nonconvulsive status epilepticus. Epilepsia. 2013;54:28-9.
24. Hirsch LJ, LaRoche SM, Gaspard N, Gerard E, Svoronos A,
Herman ST, et al. American Clinical Neurophysiology Society’s
Standardized Critical Care EEG Terminology: 2012 version. J Clin
Neurophysiol. 2013;30:1-27.
25. Sansevere AJ, Hahn CD,
Abend NS. Conventional and quantitative EEG in status
epilepticus. Seizure. 2019;68:38-45.
26. Ferlisi M,
Shorvon S. The outcome of therapies in refractory and
super-refractory convulsive status epilepticus and
recommendations for therapy. Brain. 2012;135: 2314-28.
27. Pearl PL, Vezina LG, Saneto RP, McCarter R, Molloy-Wells E,
Heffron A, et al. Cerebral MRI abnormalities associated with
vigabatrin therapy. Epilepsia 2009;50: 184-94.
28. Milh
M, Villeneuve N, Chapon F, Pineau S, Lamoureux S, Livet MO, et
al. Transient brain magnetic resonance imaging hyperintensity in
basal ganglia and brain stem of epileptic infants treated with
vigabatrin. J Child Neurol. 2009;24: 305-15.
29. Loscher
W. Mechanisms of drug resistance in status epilepticus.
Epilepsia. 2007;48:74-7.
30. Hocker S. Systemic
complications of status epilepticus– An update. Epilepsy Behav.
2015;49:83-7.
31. Glauser T, Shinnar S, Gloss D,
Alldredge B, Arya R, Bainbridge J, et al. Evidence-Based
Guideline: Treatment of Convulsive Status Epilepticus in
Children and Adults: Report of the Guideline Committee of the
American Epilepsy Society. Epilepsy Curr. 2016;16:48-61.
32. Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C,
Rowan AJ, et al. A comparison of four treatments for generalized
convulsive status epilepticus. Veterans Affairs Status
Epilepticus Cooperative Study Group. N Engl J Med.
1998;339:792-8.
33. Silbergleit R, Durkalski V,
Lowenstein D, Conwit R, Pancioli A, Palesch Y, et al.
Intramuscular versus intravenous therapy for prehospital status
epilepticus. N Engl J Med. 2012;366:591-600.
34.
Chamberlain JM, Okada P, Holsti M, Mahajan P, Brown KM, Vance C,
et al. Lorazepam vs diazepam for pediatric status epilepticus: a
randomized clinical trial. JAMA. 2014;311:1652-60.
35.
Arya R, Kothari H, Zhang Z, Han B, Horn PS, Glauser TA. Efficacy
of nonvenous medications for acute convulsive seizures: A
network meta-analysis. Neurology. 2015;85:1859-68.
36. US
Food and Drug Administrations. https://www.
accessdata.fda.gov/drugsatfda_docs/label/2019/ 211321s 000lbl
.pdf. Accessed July 31, 2019.
37. Neuralis Inc. Our
pipeline.
https://www.neurelis.com/our-pipeline/valtoco-nasal-spray.
Accessed July 31, 2019.
38. French JA, Wechsler R,
Gelfand MA, Pollard JR, Vazquez B, Friedman D, et al. Inhaled
alprazolam rapidly suppresses epileptic activity in
photosensitive partici-pants. Epilepsia. 2019.
39. Yasiry
Z, Shorvon SD. The relative effectiveness of five antiepileptic
drugs in treatment of benzodiazepine-resistant convulsive status
epilepticus: A meta-analysis of published studies. Seizure.
2014;23:167-74.
40. Dalziel SR, Borland ML, Furyk J,
Bonisch M, Neutze J, Donath S, et al. Levetiracetam versus
phenytoin for second-line treatment of convulsive status
epilepticus in children (ConSEPT): An open-label, multicentre,
randomised controlled trial. Lancet. 2019;393:2135-45.
41. Lyttle MD, Rainford NEA, Gamble C, Messahel S, Humphreys A,
Hickey H, et al. Levetiracetam versus phenytoin for second-line
treatment of paediatric convulsive status epilepticus (EcLiPSE):
A multicentre, open-label, randomised trial. Lancet.
2019;393:2125-34.
42. Bleck T, Cock H, Chamberlain J,
Cloyd J, Connor J, Elm J, et al. The established status
epilepticus trial 2013. Epilepsia. 2013;54:89-92.
43.
Rossetti AO, Milligan TA, Vulliemoz S, Michaelides C, Bertschi
M, Lee JW. A randomized trial for the treatment of refractory
status epilepticus. Neurocrit Care. 2011;14:4-10.
44.
Rosenthal ES, Claassen J, Wainwright MS, Husain AM, Vaitkevicius
H, Raines S, et al. Brexanolone as adjunctive therapy in
super-refractory status epilepticus. Ann Neurol. 2017;82:342-52.
45. Abend NS, Dlugos DJ. Treatment of refractory status
epilepticus: Literature review and a proposed protocol. Pediatr
Neurol. 2008;38:377-90.
46. Naritoku DK, Sinha S.
Prolongation of midazolam half-life after sustained infusion for
status epilepticus. Neurology. 2000;54:1366-8.
47.
Gilbert DL, Gartside PS, Glauser TA. Efficacy and mortality in
treatment of refractory generalized convulsive status
epilepticus in children: A meta-analysis. J Child Neurol.
1999;14:602-9.
48. Singhi S, Murthy A, Singhi P,
Jayashree M. Continuous midazolam versus diazepam infusion for
refractory convulsive status epilepticus. J Child Neurol.
2002;17:106-10.
49. Morrison G, Gibbons E, Whitehouse WP.
High-dose midazolam therapy for refractory status epilepticus in
children. Intensive Care Med. 2006;32:2070-6.
50.
Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of
refractory status epilepticus with pentobarbital, propofol, or
midazolam: A systematic review. Epilepsia. 2002;43:146-53.
51. Kim SJ, Lee DY, Kim JS. Neurologic outcomes of pediatric
epileptic patients with pentobarbital coma. Pediatr Neurol.
2001;25:217-20.
52. Holtkamp M, Masuhr F, Harms L,
Einhaupl KM, Meierkord H, Buchheim K. The management of
refractory generalised convulsive and complex partial status
epilepticus in three European countries: A survey among
epileptologists and critical care neurologists. J Neurol
Neurosurg Psychiatry. 2003;74:1095-9.
53. Bledsoe KA,
Kramer AH. Propylene glycol toxicity complicating use of
barbiturate coma. Neurocrit Care. 2008;9:122-4.
54.
Rossetti AO, Reichhart MD, Schaller MD, Despland PA,
Bogousslavsky J. Propofol treatment of refractory status
epilepticus: A study of 31 episodes. Epilepsia. 2004;45:757-63.
55. Parviainen I, Uusaro A, Kalviainen R, Mervaala E,
Ruokonen E. Propofol in the treatment of refractory status
epilepticus. Intensive Care Med. 2006;32:1075-9.
56. van
Gestel JP, Blusse van Oud-Alblas HJ, Malingre M, Ververs FF,
Braun KP, van Nieuwenhuizen O. Propofol and thiopental for
refractory status epilepticus in children. Neurology.
2005;65:591-2.
57. McHugh P. Acute choreoathetoid
reaction to propofol. Anaesthesia. 1991;46: 425.
58.
Enting D, Ligtenberg JJ, Aarts LP, Zijlstra JG. Total
suppression of cerebral activity by thiopental mimicking
propofol infusion syndrome: A fatal common pathway? Anesth
Analg. 2005;100:1864-5; author reply 1865.
59. Baumeister
FA, Oberhoffer R, Liebhaber GM, Kunkel J, Eberhardt J,
Holthausen H, Peters J. Fatal propofol infusion syndrome in
association with ketogenic diet. Neuropediatrics. 2004;35:250-2.
60. Hofler J, Trinka E. Intravenous ketamine in status
epilepticus. Epilepsia. 2018;59: 198-206.
61. Keros S,
Buraniqi E, Alex B, Antonetty A, Fialho H, Hafeez B, et al.
Increasing ketamine use for refractory status epilepticus in US
pediatric hospitals. J Child Neurol. 2017;32: 638-46.
62.
Rossetti AO, Logroscino G, Bromfield EB. Refractory status
epilepticus: Effect of treatment aggressiveness on prognosis.
Arch Neurol. 2005;62:1698-702.
63. An J, Jonnalagadda D,
Moura VJ, Purdon PL, Brown EN, Westover MB. Variability in
pharmacologically-induced coma for treatment of refractory
status epilepticus. PLoS. One 2018;13:e0205789.
64.
Tasker RC, Goodkin HP, Sanchez Fernandez I, Chapman KE, Abend
NS, Arya R, et al. Refractory status epilepticus in children:
Intention to treat with continuous infusions of midazolam and
pentobarbital. Pediatr Crit Care Med. 2016;17:968-75.
65.
Alvarez V, Lee JW, Westover MB, Drislane FW, Novy J, Faouzi M,
et al. Therapeutic coma for status epilepticus: Differing
practices in a prospective multicenter study. Neurology.
2016;87:1650-9.
66. Sutter R, Marsch S, Fuhr P, Kaplan
PW, Ruegg S. Anesthetic drugs in status epilepticus: risk or
rescue? A 6-year cohort study. Neurology. 2014;82:656-64.
67. Arya R, Rotenberg A. Dietary, immunological, surgical,
and other emerging treatments for pediatric refractory status
epilepticus. Seizure. 2019;68:89-96.
68. Likhodii SS,
Serbanescu I, Cortez MA, Murphy P, Snead OC, 3rd, Burnham WM.
Anticonvulsant properties of acetone, a brain ketone elevated by
the ketogenic diet. Ann Neurol. 2003;54:219-26.
69. Rho
JM, Anderson GD, Donevan SD, White HS. Acetoacetate, acetone,
and dibenzylamine (a contaminant in l-(+)-beta-hydroxybutyrate)
exhibit direct anticonvulsant actions in vivo. Epilepsia.
2002;43:358-61.
70. Arya R, Peariso K, Gainza-Lein M,
Harvey J, Bergin A, Brenton JN, et al. Efficacy and safety of
ketogenic diet for treatment of pediatric convulsive refractory
status epilepticus. Epilepsy Res. 2018;144:1-6.
71.
Legriel S, Lemiale V, Schenck M, Chelly J, Laurent V, Daviaud F,
et al. Hypothermia for neuroprotection in convulsive status
epilepticus. N Engl J Med. 2016;375:2457-67.
72.
Mirsattari SM, Sharpe MD, Young GB. Treatment of refractory
status epilepticus with inhalational anesthetic agents
isoflurane and desflurane. Arch Neurol. 2004;61:1254-9.
73. Fugate JE, Burns JD, Wijdicks EF, Warner DO, Jankowski CJ,
Rabinstein AA. Prolonged high-dose isoflurane for refractory
status epilepticus: is it safe? Anesth Analg. 2010;111:1520-4.
74. Ikeda KM, Connors R, Lee DH, Khandji AG, Claassen J,
Young GB. Isoflurane use in the treatment of super-refractory
status epilepticus is associated with hippocampal changes on
MRI. Neurocrit Care. 2017;26:420-7.
75. Ferro MA, Chin
RF, Camfield CS, Wiebe S, Levin SD, Speechley KN. Convulsive
status epilepticus and health-related quality of life in
children with epilepsy. Neurology. 2014;83:752-7.
76.
Sahin M, Menache CC, Holmes GL, Riviello JJ. Outcome of severe
refractory status epilepticus in children. Epilepsia.
2001;42:1461-7.
77. Neligan A, Shorvon SD. Frequency and
prognosis of convulsive status epilepticus of different causes:
A systematic review. Arch Neurol. 2010;67:931-40.
78.
Pujar SS, Neville BG, Scott RC, Chin RF, North London Epilepsy
Research N. Death within 8 years after childhood convulsive
status epilepticus: a population-based study. Brain.
2011;134:2819-27.
79. Pujar SS, Martinos MM,
Cortina-Borja M, Chong WKK, De Haan M, Gillberg C, et al.
Long-term prognosis after childhood convulsive status
epilepticus: A prospective cohort study. Lancet Child Adolesc
Health. 2018;2: 103-111.
80. Sahin M, Menache CC, Holmes
GL, Riviello JJ, Jr. Prolonged treatment for acute symptomatic
refractory status epilepticus: outcome in children. Neurology.
2003;61:398-401.
81. Maytal J, Shinnar S, Moshe SL,
Alvarez LA. Low morbidity and mortality of status epilepticus in
children. Pediatrics. 1989;83:323-31.
82. Gurcharran K,
Grinspan ZM. The burden of pediatric status epilepticus:
Epidemiology, morbidity, mortality, and costs. Seizure
2019;68:3-8.
83. Jafarpour S, Stredny CM, Piantino J,
Chapman KE. Baseline and outcome assessment in pediatric status
epilepticus. Seizure. 2019;68:52-61.
84. Martinos MM,
Pujar S, Gillberg C, Cortina-Borja M, Neville BGR, De Haan M, et
al. Long-term behavioural outcomes after paediatric convulsive
status epilepticus: A population-based cohort study. Dev Med
Child Neurol. 2018;60: 409-16.
85. DeLorenzo RJ, Hauser
WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A
prospective, population-based epidemiologic study of status
epilepticus in Richmond, Virginia. Neurology. 1996;46:1029-35.
86. Sadarangani M, Seaton C, Scott JA, Ogutu B, Edwards T,
Prins A, et al. Incidence and outcome of convulsive status
epilepticus in Kenyan children: A cohort study. Lancet Neurol.
2008;7:145-50.
|
|
|
 |
|
