|
Clinico pathological conference |
|
|
Indian Pediatr 2013;50: 316-320 |
 |
Neonatal Cholestasis with Ductal Paucity and
Steatosis
|
|
Rajeev Khanna, Seema Alam, *Archana Rastogi and *Chhagan Bihari Sharma
From the Departments of Pediatric Hepatology and
*Pathology, Institute of Liver and Biliary Sciences, D-1,
Vasant Kunj, New Delhi, India.
Correspondence to: Rajeev Khanna, Department of
Pediatric Hepatology, Institute of Liver and Biliary Sciences, D-1,
Vasant Kunj, New Delhi 110 070.
Email:
[email protected]
|
|
Clinical Protocol
History: A 3.5-months-old male presented to us
with small bowel type of diarrhea and recurrent non-bilious vomiting
since birth; jaundice since 2.5 months of age; swelling all over body,
clay colored stools, fever, cough and respiratory difficulty since 3
months of age, and refusal to feed from 2 days prior to admission. He
also had prolonged bleeding at the site of intramuscular injection.
There was no history of abnormal movements or altered sensorium.
Investigations previously done at another center revealed severe anemia
(Hb 5.7 g/dL), coagulopathy (prothrombin time 1 min) and lower
respiratory tract infection, for which he had received three units of
whole blood transfusion, vitamin K and parenteral antibiotics, and
subsequently referred to our center.
He was second in birth order of a third degree
consanguineous union, born full term vaginally with birth weight of 3.4
kg and uneventful perinatal and antenatal periods. His 21 months old
elder brother had normal development with no similar history.
Clinical examination: On admission, he was
afebrile with mild respiratory distress and normal cardiovascular
status. He had pallor, icterus, pedal edema, angular cheilosis,
glossitis and perianal excoriation. There was no evidence of skin or
mucosal bleeds, cyanosis, clubbing, facial dysmorphism, lymphadenopathy,
cataract, coloboma, posterior embryotoxon, chorioretinitis or cherry red
spot. There were no features of xerophthalmia or rickets. His stools
were persistently acholic. He had severe malnutrition with weight Z-score
–2.37, length Z-score –3.52 and head circumference Z-score
–1.53. His abdomen was distended with ascites and parietal edema, liver
was palpable 6 cms below costal margin, firm in consistency with smooth
surface (span 9 cms) and spleen 4 cms below costal margin. Both lung
fields were clear on auscultation. There was no murmur or cardiomegaly.
Child was lethargic and irritable corresponding to hepatic
encephalopathy stage 1-2. There were no meningeal signs, features of
raised intracranial tension or neurological deficit.
Laboratory investigations: His hematological and
biochemical profile during hospital stay is mentioned in Table
I. Peripheral blood smear revealed normocytic hypochromic anemia.
Renal function tests including electrolytes were normal. Urine was
positive for non-glucose reducing substance (NGRS) but with no evidence
of proteinuria. Stool was positive for fat globules. There was no
fasting or post-prandial hypoglycemia. Triglycerides were high (509 mg/dL)
while total cholesterol was normal.
TABLE I Investigations During Hospital Stay
|
Parameter |
Day 1 |
Day 8 |
Day 20 |
Day 27 |
|
Hemoglobin (g/dL) |
10.3 |
7.4 |
9.8 |
8.0 |
|
Total leukocyte count (mm3) |
18200 |
25400 |
23400 |
20000 |
|
Differential leukocyte count (%) |
P36 L53 |
P43 L53 |
P51 L42 |
P85 L08 |
|
Platelet (cells/ mm3 X 1000) |
165 |
180 |
216 |
155 |
|
Bilirubin (Total/direct, mg/dL) |
3.5/2.1 |
7.2/4.2 |
8.0/4.6 |
11.7/6.5 |
|
Aspartate aminotransferase (IU/L) |
172 |
100 |
197 |
225 |
|
Alkaline aminotransferase (IU/L) |
67 |
50 |
53 |
53 |
|
Alkaline phosphatase (IU/L) |
94 |
70 |
103 |
107 |
|
Gamma glutamyl transpeptidase (IU/L) |
170 |
89 |
102 |
141 |
|
Protein (g/dL) |
3.0 |
5.8 |
5.2 |
5.9 |
|
Albumin (g/dL) |
1.0 |
4.0 |
2.9 |
3.0 |
|
Prothrombin time (Test/control) |
14.2/12 |
16.6/12 |
12.2/12 |
11.9/12 |
|
International normalized ratio |
1.2 |
1.4 |
1.0 |
0.9 |
|
Ammonia (µg/dL) |
203 |
65 |
181 |
118 |
Imaging: Chest and spine radiographs were normal.
Abdominal sonogram showed enlarged echogenic liver, no dilatation of
intrahepatic biliary radicles; gall bladder was contracted in the
fasting state, common bile duct (CBD) was visualized with no evidence of
triangular cord sign; spleen and kidneys were normal; there was no
adrenal calcification. Echocardiography revealed structurally normal
heart.
Course During Hospital Stay
Child was started on parenteral antibiotics,
urso-deoxycholic acid (UDCA), multivitamin supplements, medium chain
triglycerides (MCT) based feeds and albumin infusion. Packed red cells
were transfused. In view of high bilirubin, positive urine NGRS,
vomitings and irritability, possibility of Galactosemia was kept and he
was shifted to lactose free formula. Subsequently his
Galactose-1-phosphate uridyl transferase (GALT) assay came out to be
normal (15.5 g/dL; normal 15-45 g/dL), which could be false negative in
this setting with recent (within 3 months) blood transfusions. Meanwhile
other workup for intrahepatic cholestasis was done, which revealed
normal alpha-fetoprotein for age (1135 ng/mL), normal urinary
succinylacetone and normal serum alpha-1 antitrypsin (A1AT) (2.2 g/L,
normal 0.9-2.0 g/L). His arterial blood gas analysis revealed normal pH
and lactate. Arterial ammonia was high (normal <60 µg/dL). Urine and
plasma tandem mass spectrometry (TMS) did not reveal presence of
abnormal metabolite for aminoacidurias, organic acidurias, and urea
cycle, fatty-acid oxidation or respiratory chain defects. He was started
on oral sodium benzoate for hyperammonemia.
Subsequently, he developed lower respiratory tract
infection with polymorphonuclear leukocytosis. Anti-biotics were
changed, although blood and urine cultures were sterile. Immunoglobulin
profile was normal and ELISA for HIV was negative. Chest findings
improved transiently and a liver biopsy was undertaken. Magnetic
resonance cholangiopancreatography (MRCP) and upper gastrointestinal
endoscopy were planned, but the procedures were deferred considering his
poor respiratory condition.
Chest X-ray showed persistence of infiltrates
with development of bronchiectatic changes. Sweat chloride levels were
high on two occasions (75.7 mmol/L and 70 mmol/L). Mutational analysis
revealed heterozygous state for delta F508 mutation. He was started on
3% saline nebulisations and pancreatic enzyme supplements.
Lactose-containing feeds were restarted and density was increased by
adding puffed-rice powder. This was followed by improvement in stool
frequency and consistency, and resolution of steatorrhea. His
respiratory status improved and weight gain started. In view of high
bilirubin, persistent hypoalbuminemia and increasing liver size (10 cm
below costed margin) with Pediatric end-stage liver disease (PELD) score
of 20, the option of liver transplantation was given to the family and
evaluation was initiated. He was discharged in a stable condition on Day
36 of hospitalization, but he died due to recurrence of pneumonia at 5
months of age at his native place. Autopsy was not conducted.
Unit’s diagnosis: Neonatal cholestasis with
acholic stools (Ductal paucity); with Severe protein energy malnutrition
(wasting and stunting), anemia, anasarca; with chronic diarrhea and
steatorrhea; with Pneumonia.
Discussion on clinical protocol: The clinical and
laboratory information in the child suggested neonatal cholestasis.
Table II provides clinical and laboratory pointers to
establish the diagnosis in a cholestatic infant. Passage of pigmented
stools for first three months, absence of history of hemolysis or total
parenteral nutrition, sonographic visualization of gall-bladder and CBD,
with no evidence of sludge in gall bladder or CBD or dilatation of
intrahepatic biliary radicles (IHBRD), and subsequent liver biopsy
findings excluded biliary atresia, choledochal cyst and inspissated bile
duct syndrome. Although MRCP could not be done but liver biopsy did not
suggest neonatal sclerosing cholangitis [1-3].
TABLE II Key Clinical and Laboratory Pointers to Diagnosis of a Cholestatic Infant
|
Acholic stools |
Biliary atresia, Choledochal cyst, Neonatal sclerosing
cholangitis, inspissated bile duct syndrome, Spontaneous
perforation of bile duct, Ductal paucity (PILBD) |
|
Sick infant |
MLD (Galactosemia, Tyrosinemia, Neonatal hemochromatosis, FATMO
defects), Sepsis, Urinary tract infection, TORCH infections |
|
Pruritus |
PFIC types 1, 2 and 3, PILBD (Alagille’s syndrome and Non-syndromic) |
|
Ascites |
MLD (Galactosemia, Tyrosinemia, Neonatal hemochromatosis), Late
presentation of biliary atresia (>6 mo), End-stage liver disease
due to any cause, Spontaneous perforation of bile duct |
|
Dysmorphism
|
Alagille’s, Down’s syndrome, Williams syndrome, Peroxisomal
defects, TORCH infections |
|
Cardiac defect or murmur |
Alagille’s syndrome (Peripheral pulmonary artery stenosis),
Congenital rubella (Patent ductus arteriosus, Tetralogy of
Fallot, Ventricular septal defect), Biliary atresia (VSD, Atrial
septal defect) |
|
Eye findings |
Galactosemia (Cataract), Alagille’s syndrome (Posterior
embryotoxon), TORCH infections (Chorioretinitis), Niemann-Pick
disease type C (Cherry red spot)
|
|
Vertebral defects |
Alagille’s syndrome |
|
Lymphedema |
Aagenes syndrome |
|
Rickets + Renal tubular acidosis |
Galactosemia, Tyrosinemia |
|
Low GGT
|
Progressive familial intrahepatic cholestasis (PFIC) types 1 &
2, Bile acid synthetic defects |
|
Hyperammonemia |
NICCD, Advance liver failure due to any cause |
|
Hypoglycemia |
MLD (Galactosemia, Tyrosinemia, Neonatal hemochromatosis, FATMO
defects, Advance liver failure due to any cause |
Paucity of intralobular bile ducts (PILBD, or ductal
paucity) and progressive familial intrahepatic cholestasis (PFIC) are
among the common intrahepatic causes of cholestasis (Table III).
This was a full-term baby with no facial dysmorphism, heart murmur,
vertebral or heart defect, posterior embryotoxon, microcephaly,
cataract, chorioretinitis, hypoglycemic spells or skin rash, his A1AT
level was normal; thus excluding common causes of ductal paucity [4].
The high GGT disfavored the diagnosis of PFIC types 1 and 2 [5]. Cystic
fibrosis (CF) was suggested by the infant’s phenotype, which was later
confirmed by sweat chloride testing and mutational analysis [6].
TABLE III Key Histopathological Pointers in Liver Biopsy to Diagnosis of a Cholestatic Infant
|
Cholestasis (hepatocellular and canalicular bile stasis), bile
plugs, ductular proliferation
|
Biliary atresia, extrahepatic obstruction (choledochal cyst,
inspissated bile syndrome), bile-acid synthetic defects, PFIC
type 1 and 3 |
|
Portal fibrosis with early cirrhosis |
Biliary atresia, choledochal cyst, Metabolic disorders (Galactosemia,
Tyrosinemia, Neonatal hemochromatosis, A1AT deficiency), PFIC
type 2 |
|
Lobular and portal inflammation, giant-cell transformation,
extramedullary hematopoiesis |
Idiopathic neonatal hepatitis, TORCH infections, PFIC type 2,
MLD |
|
Ductal paucity
|
Alagille’s syndrome, Non-syndromic PILBD (α1AT
deficiency, CF, Down’s syndrome, Williams syndrome, Peroxisomal
disorders, Prematurity, TORCH infections, hypopituitarism, PFIC
types 1 and 2) |
|
Steatosis |
MLD (Galactosemia, Hereditary fructose intolerance, Tyrosinemia,
a1AT deficiency, CF, NICCD, Peroxisomal disorders, FATMO
defects)
|
|
PAS-positive diastase resistant globules in periportal
hepatocytes |
α1AT deficiency |
|
PFIC = Progressive familial intrahepatic cholestasis, A1AT =
Alpha-1 antitrypsin deficiency, TORCH = Toxoplasma, Rubella,
Cytomegalovirus, Herpes, Others, MLD = Metabolic liver disease,
PILBD = Paucity of intralobular bile ducts, CF = Cystic
fibrosis, NICCD = Neonatal intrahepatic cholestasis caused by
citrin deficiency, FATMO = Fatty-acid transport and
mitochondrial oxidation.
|
Among the metabolic liver disease, Galactosemia was
considered in view of vomitings, irritability, failure to thrive,
positive NGRS, chest infection and deteriorating liver functions;
although, acholic stools, absence of hypoglycemia, non-response to
galactose-free formula were against this possibility. Absence of liver
failure with normal urinary succinylacetone excluded Tyrosinemia.
Hyperammonemia and normal lactate with evidence of liver dysfunction
suggested a possibility of neonatal intrahepatic cholestasis caused by
citrin deficiency (NICCD), but the disorder was excluded on TMS [5].
Pathology Protocol
Liver tissue comprised of ten portal areas.
Hematoxylin and eosin staining revealed marked acinar disarray with
swollen hepatocytes filled with marked macrovesicular steatosis (90%).
There was cytoplasmic rarefaction with pseudoacinar transformation,
along with hepatocellular and canalicular cholestasis, and formation of
cholestatic rosettes. Few hepatocytes showed glycogenated nuclei.
Sinusoids were compressed. Portal areas displayed minimal mixed
inflammation with absence of bile ducts in 7 out of 10 tracts. Masson’s
Trichrome staining showed mild periportal fibrosis. Periodic acid
Schiff’s (PAS) staining alone and with prior diastase digestion didn’t
reveal any cytoplasmic inclusions (Fig. 1).
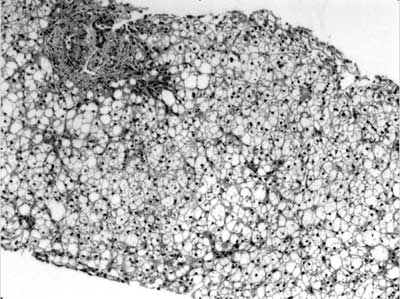 |
|
Fig. 1 Liver biopsy (Hematoxylin and
Eosin stain, 100X) showing acinar disarray, distended
hepatocytes with marked macrovesicular steatosis. Portal tract
reveals portal fibrosis with periportal extension, and absence
of bile duct.
|
Pathological diagnosis: Paucity of intralobular
bile ducts; Macrovesicular steatosis.
Discussion on Pathology Protocol: Liver biopsy
findings in this 3.5 months old male infant suggest ductal paucity with
marked steatosis. Table III shows important
histopathlogical pointers in liver biopsy of a cholestatic infant which
may provide clue to diagnosis. Ductal paucity or PILBD is a histological
term defined as number of bile ducts to portal tracts (BD/PT) ratio of
less than 0.4 as mentioned for adults (normal range in children 0.9-1.8)
commented in a biopsy containing at least 10 portal tracts [3, 7]. The
concerned infant had ductal paucity (BD/PT ratio 0.3). Steatosis with
ductal paucity at this young age pointed towards two differentials -
a1AT
deficiency and CF [4].
Liver biopsy findings of AIAT deficiency are highly
variable and include portal and lobular inflammation, necrosis, ductal
paucity, ductular proliferation, portal fibrosis, cirrhosis and variable
steatosis. The characteristic histopathological hallmark in the form of
PAS positive, diastase resistant eosinophilic inclusions comprised of
retained A1AT globules in periportal hepatocytes and bile duct
epithelial cells are usually seen in infants older than 3 months, which
increase in number subsequently [8].
Liver pathology in CF in young infants usually
includes steatosis, inflammation, paucity of intralobular bile ducts,
ductular proliferation, portal inflammation, portal expansion, fibrosis,
cirrhosis and cholestasis [7, 9-11]. The pathognomonic lesion of
CF-related liver disease CFLD) is focal biliary cirrhosis, which
develops due to blockage of biliary ductules secondary to viscid
secretions [7]. Focal changes are seen in 11% of infants, 26% of those
dying at one year; and in more than 70% of adults, and they later on
progress to multilobular biliary cirrhosis with subsequent portal
hypertension. Steatosis is seen in 23-67% and is panlobular
macrovesicular type and may occur without focal biliary cirrhosis [7, 9,
12].
Final diagnosis: Cystic fibrosis with lung
and liver involvement (pneumonia, bronchieactasis, cholestasis with
liver dysfunction); Severe malnutrition, anemia.
Open Forum
Cystic fibrosis (CF) is a rare cause of neonatal
cholestasis (0.6-0.7%) [10, 11]. Suggestive phenotype and genotype with
high sweat chloride levels in our infant fulfilled the diagnostic
criteria [6]. Neonatal or infantile cholestasis is a known but unusual
manifestation of CF [9]. Overall, prevalence of CF in the West is ~1 in
3500 newborns [6]. In India, the estimated prevalence is low, but the
diagnosis is now getting increasingly recognized due to awareness and
availability of diagnostic tests [13].
Cystic fibrosis-related liver disease (CFLD) is seen
in 55-70% of cases with CF with median age at diagnosis of 12 years and
is associated with severe malnutrition, early death and risk for lung
transplantation [9, 14]. Portal hypertension is predicted by presence of
fibrosis, and occurs at a young age with severe fibrosis [12]. Risk of
CFLD in CF is around 1.8% per 100 person-years and is more with male
gender, younger age, history of meconium ileus, or presence of severe
mutations [15].
Infants with CF have evidence of biochemical CFLD in
more than 50% of cases, but such mild biochemical abnormalities revert
to normal in most of them [9]. Presentation as neonatal cholestasis is
rare as per two older series describing 21 cases; three of which
mimicked biliary atresia. Jaundice, hepatomegaly, hypoalbumi-nemia,
meconium ileus, acholic stools, and chest symptoms were the presenting
features. Jaundice cleared within a median duration of 7.4 months.
Clinical or biochemical CFLD and cirrhosis developed in 13 and 4 cases,
respectively [10, 11]. One Indian series has reported one CF among 410
neonatal cholestasis cases (0.002%) from a tertiary referral centre, but
the exact details are lacking [16].
Management of CFLD is aimed at nutritional support
and UDCA, with subsequent focus on management of cirrhosis and its
complications [17]. Liver transplantation is done either alone or
combined with lung and is shown to have comparable outcomes [18].
Indications for transplantation are decided on
basis of model for end-stage liver disease (MELD) or pediatric end-stage
liver disease (PELD) score, giving additional points if forced
expiratory volume in 1 second is less than 40% [19]. An algorithm for
consideration of patients for LT or LLT in CFLD has been proposed [18].
In conclusion, cystic fibrosis, CF, although a rare
cause of neonatal cholestasis, should be considered in the work-up of an
infant with cholestasis after the common etiologies have been excluded.
Contributors: RK and SA designed the
format of clinicopathological conference and revised the manuscript for
intellectual content. RK will act as the guarantor. AR and CBS conducted
pathology protocol and provided pathology discussion. SA and AR drafted
the final clinical and pathology discussion. All authors participated in
open forum.
Funding: None; Competing interests: None
stated.
References
1. Bruyne RD, Biervliet SV, Velde SV, Winckel MV.
Clinical practice - Neonatal cholestasis. Eur J Pediatr.
2011;170:279-84.
2. Guideline for the Evaluation of Cholestatic
Jaundice in Infants: Recommendations of the North American Society for
Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr
Gastroenterol Nutr. 2004;39:115-28.
3. Suchy FJ. Anatomy, histology, embryology,
developmental anomalies, and pediatric disorders of the biliary tract.
In: Feldman M, Friedman LS, Brandt LJ (eds). Sleisenger and Fordtran’s
Gastrointestinal and Liver Disease, Pathophysiology/Diagnosis/Management,
Ninth edition. Saunders, an imprint of Elsevier Inc., Philadelphia,
2010; p 1045-66.
4. Sinha J, Magid M S, VanHuse C, Thung S N, Suchy F,
Kerkar N. Bile duct paucity in infancy. Semin Liver Dis. 2007;27:319-23.
5. Hansen K, Horslen S. Metabolic liver disease in
children. Liver transplantation. 2008;14:713-33.
6. Farrell PM, Rosenstein BJ, White TB, Accurso FJ,
Castellani C, Cutting GR, et al. Guidelines for Diagnosis of
Cystic Fibrosis in Newborns Through Older Adults: Cystic Fibrosis
Foundation Consensus Report. J Pediatr. 2008;153:S4–S14.
7. Russo P, Rand EB, Haber BA, Loomes KM. Diseases of
the biliary tree in infancy and childhood. In: Russo P, Ruchelli
ED, Picolli DA (eds). Pathology of Pediatric
Gastroenterointestinal and Liver disease 2004, Springer-Verlag New York,
Inc., p 203-36.
8. Teckman JH. Alpha-1 antitrypsin deficiency in
childhood. Semin Liver Dis. 2007:27:274-81.
9. Lindblad A, Glaumann H, Strandvik B. Natural
history of liver disease in cystic fibrosis. Hepatology. 1999;30:1151-8.
10. Lykavieris P, Bernard O, Hadchouel M. Neonatal
cholestasis as the presenting feature in cystic fibrosis. Arch Dis
Child. 1996;75:67-70.
11. Shapira R, Hadzic N, Francavilla R, Koukulis G,
Price JF, Mieli-Vergani G. Retrospective review of cystic fibrosis
presenting as infantile liver disease. Arch Dis Child. 1999;81:125-8.
12. Lewindon PJ, Shepherd RW, Walsh MJ, Greer RM,
Williamson R, Pereira TN, et al. Importance of hepatic fibrosis
in cystic fibrosis and the predictive value of liver biopsy. Hepatology.
2011;53:193-201.
13. Kabra SK, Kabra M, Lodha R, Shastri S, Ghosh M,
Pandey RM, et al. Clinical profile and frequency of delta F 508
mutation in Indian children with cystic fibrosis. Indian Pediatr.
2003;40:612-9.
14. Chryssostalis A, Hubert D, Coste J, Kanaan R,
Burgel PR, Desmazes-Dufeu N, et al. Liver disease in adult
patients with cystic fibrosis: A frequent and independent prognostic
factor associated with death or lung transplantation. J Hepatol.
2011;55:1377-82.
15. Bhardwaj S, Canlas K, Kahli C, Temkit M,
Molleston J, Ober M, et al. Hepatobiliary abnormalities and
disease in cystic fibrosis: epidemiology and outcomes through adulthood.
J Clin Gastroenterol. 2009;43:858-64.
16. Arora NK, Arora S, Ahuja A, Mathur P, Maheshwari
M, Das MK, et al. Alpha-1 antitrypsin deficiency in children with
chronic liver disease in North India. Indian Pediatr. 2010;47:1015-23.
17. Debray D, Kelly D, Houwen R, Strandvik B, Colombo
C. Best practice guidance for the diagnosis and management of cystic
fibrosis-associated liver disease. J Cyst Fibros. 2011;10:S29-S36.
18. Arnon R, Annunziato RA, Miloh T, Padilla M,
Sogawa H, Batemarco L, et al. Liver and combined lung and liver
transplantation for cystic fibrosis: Analysis of the UNOS database.
Pediatr Transplantation. 2011:15:254-64.
19. Horslen S, Sweet S, Gish RG, Shepherd R. Model
for end-stage liver disease (MELD) exception for cystic fibrosis. Liver
Transplantation. 2006;12:S98–S99.
|
|
|
 |
|
