|
|
Case Reports Indian Pediatrics 2007;44:223-225 |
||
|
Fanconi-Bickel Syndrome |
||
|
Sunil Karande From the Developmental Clinic, Department of Pedia-trics, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai 400 022, Maharashtra, India. Correspondence to: Dr. Sunil Karande, Flat 24,
Joothica, 5th Floor, 22A, Naushir Bharucha Road,, Mumbai 400 007,
Maharashtra, India. Manuscript received: June 27, 2006; Initial review
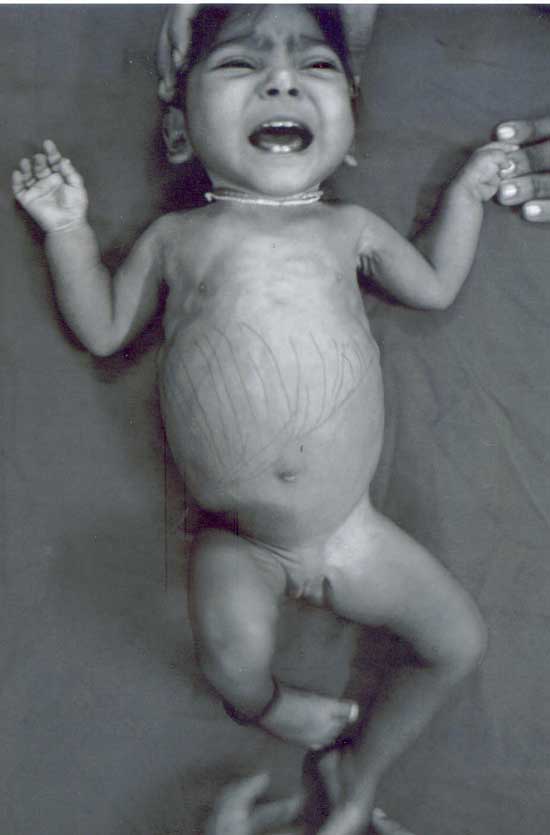
completed: July 7, 2006; Abstract: We present here the first case of Fanconi-Bickel syndrome, a rare type of glycogen storage disease, from India. A 17-month-old female child presented with severe growth retardation and abdominal distention. Clinical examination revealed a "doll-like" face, massive hepatomegaly, and rickets. Laboratory investigations confirmed severe hypophosphatemic rickets and proximal renal tubular dysfunction. Liver biopsy showed glycogen accumulation in the hepatocytes. Key words: Glycogen storage disease type XI, Glycogenosis, Hepatomegaly. Fanconi-Bickel syndrome (FBS) or Hepato-renal Glycogenosis with Renal Fanconi Syn-drome or Glycogen Storage Disease Type XI is an extremely rare disorder of carbohydrate metabolism which was first described by Fanconi and Bickel in 1949(1). Since then around 112 cases have been published in world literature(2,3). It is an autosomal recessive disease for which an enzymatic defect has still not been identified(2,3). The disease is characterized by the association of a massive liver due to glycogen accumulation; and severe hypophosphatemic rickets and marked growth retardation due to proximal renal tubular dysfunction(2,3).- Case Report A 17-month-old female child, first born of a third degree consanguineous marriage and belonging to the Muslim community, presented with failure to thrive and abdominal distention. There was no history of any antenatal problems or any medical illness in the mother. She was a preterm (33 weeks gestation, birth weight 2.1 kg) baby delivered by emergency cesarean section for meconium stained liqor. There was history of delayed cry after birth. There was no history of seizures, icterus, or cataract. The child had growth retardation (head circumference 36 cm, length 57 cm, weight 4 kg, all <3rd percentile), a "doll-like" (or "moon-shaped") face, and clinical manifestations of rickets (Fig. 1). The liver span was 14 cm. Rest of the systemic examination was normal. The developmental age of the child was seven months.
Investigations revealed normal serum calcium (9 mg/dL), reduced serum phosphorus(2.1 mg/dL, normal 2.7-4.5), and markedly elevated serum alkaline phosphatase (2850 U/L, normal 145-420 U/L) levels. Blood glucose level two hours after feed was elevated (124 mg/dL, normal 60-100). Liver function tests (serum proteins, bilirubin, ALT, AST and prothrombin time) were normal. Serum cholesterol (217 mg/dL, normal 45-182 mg/dL) and triglycerides (268 mg/dL, normal 32-99 mg/dL) were elevated. Serum HDL cholesterol and LDL cholesterol levels were normal. Blood urea nitrogen and serum creatinine, uric acid and lactate levels were normal. Arterial blood gas examination revealed metabolic acidosis (pH 7.2, bicarbonate 15.9 mmol/L). Serum sodium and potassium levels were normal. Urinary pH was 6.4 (normal 4.5-8), with 4+glucosuria, 1+proteinuria and generalized aminoaciduria. X-ray of the wrist showed active rickets. Abdominal ultrasono-graphy showed massive hepatomegaly with coarse echotexture. The kidney size (6.6 cm) was above the height-corrected normal value of 5.1 cm(4). Liver biopsy showed marked accumulation of glycogen in the hepatocytes. A diagnosis of Fanconi-Bickel syndrome was made. The child was commenced on treatment with oral vitamin D2 supplementation (2000 IU/kg/24 hr as a single daily dose) and oral phosphate supplementation (0.6 g/24 hr) given as Joulie solution (4 mL given every 4 hr, 5 times daily) for the hypophosphatemic rickets; and oral bicarbonate supplementation (6 mEq/kg/24 hr) given as sodamint tablets (3 tablets three times a day) to correct the metabolic acidosis(2,3). The mother was advised to give the child frequent and small meals with adequate caloric intake and to delete milk from the child’s diet(2,3). The child has followed to date up to 22 months of age and still has growth retardation and other findings of FBS. Discussion FBS is an extremely rare but distinct clinical entity(2,3). This disorder has been reported from all parts of Europe, Turkey, Israel, Arabian countries of the Near East and North Africa, Japan, and North America(2,3). The affected child presents between age of 3 to 10 months with failure to thrive, "doll-like" face, severe hypophosphatemic rickets, and massive hepatomegaly(2,3). Proximal renal tubular dysfunction with glucosuria, phosphaturia, generalized aminoaciduria, bicarbonate wasting, and hypophosphatemia are characteristic findings(2,3). By two years of age enlarged kidneys are noticeable clinically(5). Fasting hypoglycemia, hyperglycemia and hypergalactosemia in the post absorptive state indicating an impaired utilization of these two monosaccharides, and hyperlipidemia may be present(2,3). Till today the precise pathophysiological mechanisms for the impaired glucose and galactose utilization often detected in FBS are far from clear(2). There is no specific therapy. The administration of uncooked cornstarch has been reported to be beneficial on metabolic control and for promoting growth(6). Puberty is delayed and after onset of puberty the hepatomegaly has been documented to recede(2,3). Glomerular filtration rate remains normal or is slightly decreased(2,3,5). Overall prognosis for survival to adulthood seems to be favorable; in addition to Fanconi and Bickel’s original patient at least two more patients have reached adulthood in stable condition(2). In recent times FBS has been diagnosed in neonatal screening programs measuring blood galactose in Guthrie test cards(7). Also, recent genetic research has indicated that FBS is a single gene disease and is caused by defects in the facilitative glucose transporter 2 (GLUT2) gene on chromosome 3q26.1-26.3 which encodes for the glucose transporter protein 2 expressed in hepatocytes, pancreatic beta-cells, enterocytes, and renal tubular cells(8,9). However subsequent research has challenged this notion of FBS being a single disease(10). The only differential diagnosis of Fanconi-Bickel syndrome is Von Gierke disease (or Hepatorenal Glycogenosis or Glycogen Storage Disease Type I a) which is also characterized by growth retardation, "doll-like" face, massive hepatomegaly and renomegaly(2,3). However, there is no proximal renal tubular dysfunction and hypophosphatemic rickets does not develop(2,3). Also, Von Gierke disease is characterized by defective glucose-6-phosphatase enzyme activity; severe episodes of hypoglycemia; elevated serum lactate, cholesterol, triglycerides and uric acid levels; and bleeding episodes(2,3). Acknowledgement We thank our Dean, Dr. M.E. Yeolekar, for granting us permission to publish this case report. Contributors: SK diagnosed and managed the case, reviewed the literature, drafted the article and will act as the guarantor for the article. NK managed the case, helped in reviewing the literature, discussed core ideas and edited the manuscript; MK supervised management of the case, discussed core ideas and edited the manuscript. Funding: None. Competing interests: None.
| ||
|
References | ||
|
![]()