Primary
immunodeficiency disorders, now known as Inborn
errors of immunity, are a group of rare diseases
affecting different arms of immune system. With the
increased use of next generation sequencing, novel
genes are being identified that have broadened our
understanding of different clinical and
immunological phenotypes. International Union of
Immunological Societies have been updating the
genetic causes for primary immunodeficiency
disorders since 1970 [1]. Since 2013, the expert
committee has started updating the phenotypic
classification for the ease of practicing physicians
[2]. In the last update, the inborn errors of
immunity were classified in to nine categories, some
of which were sub-classified in two categories based
on severity of the disease [2]. The latest update
has ten categories with a new category of bone
marrow failures and 65 new disorders with total
number of disorders reaching 430 (Fig.1)
[1,3]. Different modes of inheritance and distinct
mechanisms like loss of function, gain of function,
haploinsufficiency and dominant negative forms
leading to different phenotypes are reported for 35
genes (Fig.2).
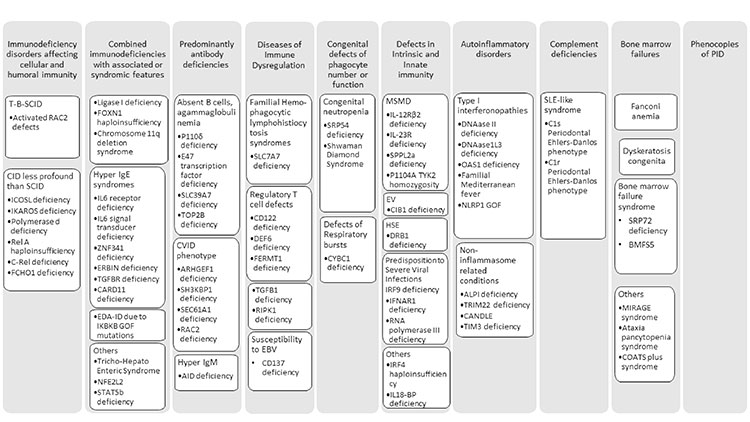
Prepared from:
SCID-Severe combined immunodeficiency
disorder, CID-Combined immunodeficiency
disorder, EDA ID-Anhidrotic
ectodermodysplasia with immunodeficiency,
CVID-Common variable immunodeficiency
disorder, MSMD- Mendelian susceptibility to
mycobacterial disease. |
| Fig. 1
New disorders included in the 2019 update. |
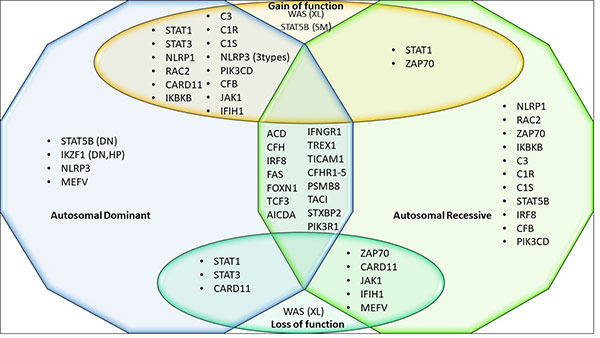
Prepared from:
XL- X linked inheritance, SM- Somatic
mutations, DN- Dominant negative, HP-
Haploinsufficiency. |
| Fig. 2
Genes with multiple modes of inheritance and
mechanisms. |
In the category of Immunodeficiencies affecting
cellular and humoral immunity, eight new genes are
added. An autosomal dominant (AD) gain of function
in RAC2 gene causes recurrent bacterial and
viral infections and may also be associated with
neutropenia and lymphoproliferation. ICOSL
deficiency patients have recurrent viral respiratory
tract infections (RTI) with slowly progressive
neutropenia and may have chronic diarrhea. IKAROS
deficiency patients present with opportunistic
infections like P.jirovecii, and
agammaglo-bulinemia. These patients are at an
increased risk to develop B cell ALL. Polymerase
deficiency patients are of short stature and had
recurrent respiratory tract and skin infections with
molluscum contagiosum and viral warts. Rel A
Haploinsufficiency patients have increased
inflammatory cytokines signaling causing chronic
mucocutaneous ulceration. c-Rel deficiency causes
increased susceptibility to opportunistic organisms
like Salmonella, Cryptobacterium, Cytomega-lovirus
and Mycobacterium tuberculosis. FCHO1 deficient
patients have failure to thrive,
lympho-proliferation with predisposition to
recurrent infections.
In Combined immunodeficiency with associated or
syndromic features twelve new disorders have been
added. LIG1 deficiency patients have growth
retardation, increased sensitivity to radiation and
sunlight with recurrent bacterial and viral
infections. FOXN1-haploinsufficient patients
also have recurrent bacterial and viral infections
with eczema, dermatitis and nail dystrophy.
Chromosome 11q23 deletion which causes Jacobsen
syndrome have growth retardation, facial
dysmorphism, warts and recurrent RTI. Seven
disorders have been added with features like Hyper
IgE syndrome due to mutations in IL6R, IL6ST,
ZNF341, ERBB2IP,TGFBR1,TGFBR2 and loss of
function forms in CARD11. Recurrent
bacterial, viral and fungal infections with
ectodermal dysplasia is also seen due to gain of
function mutations in IKBKB. Other syndromic
defects are also listed (Fig. 1). A
dominant negative form of STAT5b deficiency
with eczema and growth failure without immune
defects like the autosomal recessive (AR) form is
also added in this category.
In predominantly antibody deficiencies due to severe
reduction in all serum immunoglobulin isotypes with
profoundly decreased or absent B cells four
disorders are added. p110d deficient patient have severe bacterial
infections and autoimmune complications like
inflammatory bowel disease (IBD). AR forms of E47
transcription factor deficiency are more severe than
the AD forms and present with recurrent bacterial
infections and failure to thrive. SLC39A7
(ZIP7) deficiency present at an early age with
recurrent infections, blistering dermatosis and may
have thrombocytopenia. Hoffman syndrome due to
TOP2B deficiency present with recurrent
infections with limb anomalies and facial
dysmorphism. Four new disorders with Common variable
immunodeficiency phenotype have been described (Fig.
1). AD forms of AID deficiency causing
Hyper IgM phenotype with lymphadenopathy and
autoimmunity is also mentioned.
In diseases of immune dysregulation,
one new disorder is added in familial hemophagocytic
lymphohistiocytosis syndrome, SLC7A7 defect
leading to lysinuric protein intolerance which may
be associated with pulmonary alveolar proteinosis
and bleeding tendencies. Three new disorders with
regulatory T cell (Treg) defects are CD122
deficiency with lympho-proliferation and
autoimmunity, DEF6 deficient patient had
enteropathy, hepatosplenomegaly and cardio-myopathy
and FERMT1 deficient patients with severe
dermatosis. TGFB1 deficiency and RPIK1
defects are new causes of immune dysregulation with
colitis and CD137 deficiency a cause of increased
susceptibility to Epstein-Barr virus and
lymphoproliferation.
Two new disorders with congenital
neutropenia are SRP54 deficiency with
exocrine pancreatic deficiency and
neurodevelopmental delay and ELF1 defect an
additional cause for Shwachman-Diamond syndrome. A
new form of functional phagocytic defect due to
CYBC1 deficiency causes inflammatory
gastrointestinal symptoms with recurrent bacterial
infections.
Mendelian susceptibility to mycobacterial disease
involves three new genes namely IL12RB2,
IL23R and SPPL2A, all of which predispose
to mycobacterial and salmonella infections and a
homozygous P1104A in TYK2 gene which is
associated with increased risk of tuberculosis.
CIB1 deficiency causes increased risk of
epidermodysplasiaverruciformis (EV). IRF9
deficient patients are at an increased risk of
severe influenza infections. IFNAR1 patients
have severe disease after yellow fever and measles
vaccination. Severe varicella zoster virus
infections may be seen due to RNA polymerase III
deficiency. DBR1 deficiency can cause herpes
simplex encephalitis (HSE) along with other viral
infections of brain stem. IRF4
haploinsufficiency is included under other inborn
errors of immunity related to leukocytes presenting
as Whipple disease.
Nine new disorders with Autoinflammatory phenotype
are added in the update which include DNASE2
deficiency with features likeAcardi-Goutieres
Syndrome, DNASE1L3 deficiency causing
pediatric systemic lupus erythematosus, OAS1
deficiency causing pulmonary alveolar proteinosis
and skin rashes, an AD form of MEFV gene
mostly M694del causing familial Mediterranean fever
and NLRP1 GOF variant causing juvenile onset
recurrent respiratory papillomatosis, corneal
scarring and palmoplantar carcinoma.
Non-inflammasome related conditions included are
ALPI and TRIM22 deficiency causing IBD,
mutations in PSMG2 gene causing auto-immune
hemolytic anemia, lipodystrophy and panniculitis
with CANDLE like phenotype and T cell lymphoma
subcutaneous panniculitis-like (TIM3
deficiency).
In complement deficiency disorders GOF variants of
C1S and C1R defects causing
periodontal Ehlers-Danlos like phenotype causing
hyperpigmentation and skin fragility are included in
the new update.
To summarize, with frequent use of whole exome
sequencing and whole genome sequencing the list of
monogenic disorders will keep on increasing in times
to come. With new genetic defects being
identified,specific targeted therapies can be used
increasingly like in patients with mutations in
JAK1, STAT1, STAT3, PIK3CD, DFE6, CTLA4, LRBA,
IL12Râ2, IL23R AND IL18BP genes [1].With
clinical and immunophenotypic correlation of newer
genes being identified, our understanding of
immunology is bound to evolve.
Contributors:
BU: conceived and written the manuscript. MM revised
the manuscript for important intellectual content.
The final manuscript was approved by all authors.
Funding:
Indian Council of Medical Research (ICMR);
Competing Interests: None stated.
REFERENCES
1. Tangye SG,
Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles
C, Etzioni A, et al. Human Inborn Errors of
Immunity: 2019 Update on the Classification from the
International Union of Immunological Societies
Expert Committee. J Clin Immunol. 2020;
2. Picard C,
Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L,
Chatila T, et al. International Union of
Immunological Societies: 2017 Primary
Immuno-deficiency Diseases Committee Report on
Inborn Errors of Immunity. J Clin Immunol.
2018;38(1):96-128.
3. Bousfiha
A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila
T, et al. Human Inborn Errors of Immunity:
2019 Update of the IUIS Phenotypical Classification.
J Clin Immunol. 2020: 11; 1-16.


