Genomic testing
refers to the analysis of human DNA to detect disease-causing
variations. These variations could be chromosomal abnormalities
or single gene defects (monogenic or Mendelian disorders).
Chromo-somal abnormalities can be numerical (aneuploidy) or
structural, which include loss or gain of a large part of one or
more chromosomes, translocations, inversions and insertions.
Loss or gain of smaller regions of a chromosome, known as copy
number variations (CNV), usually involve more than one gene and
are implicated in many human diseases [1]. While chromosomal
aneuploidies are traditionally detected by karyotyping,
chromosomal microarray analysis (CMA) is now widely used to
detect chromosomal abnormalities. Next gene-ration sequencing
(NGS), which includes targeted panel testing, exome sequencing
(ES) and whole genome sequencing (WGS), has emerged as the most
powerful tool for diagnosis of monogenic disorders, which are
mostly caused by sequence variations in the coding portion of
the DNA. With technological advances, cost of these tests has
decreased drastically and they have become widely available.
This review discusses the techniques, clinical utility,
advantages and limitations of CMA and NGS.
CHROMOSOMAL MICROARRAY
CMA, otherwise known as genomic microarray, enables the study
of chromosomes at a higher resolution as compared to traditional
karyotyping. It has replaced karyotyping as the first-tier
investigation of children with intellectual disability, multiple
malformations and autism [2,3].
Principle
CMA is based on complementary hybridization of nucleotides in
the probe and target DNA. Probes are oligonucleotides, varying
in length from 25 to 70 bp, which are immobilized on a glass
slide or a chip (array) [4-7]. They are spread across the genome
at regular intervals (form the ‘backbone’ and defines the
resolution of CMA) and are usually enriched for regions of
clinical interest. They are designed to detect CNVs or single
nucleotide polymorphisms (SNPs) or both. A CNV is a segment of
DNA, which is 1kb or more, and has a variable copy number (extra
or less) compared to reference genome [8]. SNPs are the most
common genetic variations found in a population across the human
genome. Genotyping of millions of SNPs across the genome
provides information on alleles and their copy numbers, in
addition to mosaicism, uniparental disomy, triploidy and regions
of homozygosity. The different types of oligo array platforms
include comparative genomic hybridization arrays (array CGH) and
SNP arrays
(Fig. 1a and 1b). Most commercially
available platforms are hybrid arrays and contain
oligonucleotide probes for detecting both CNVs and SNPs. Array
design can be targeted (for specific regions of interest), whole
genome (evaluates entire genome) or a combination of whole
genome and targeted (most commercially available platforms).
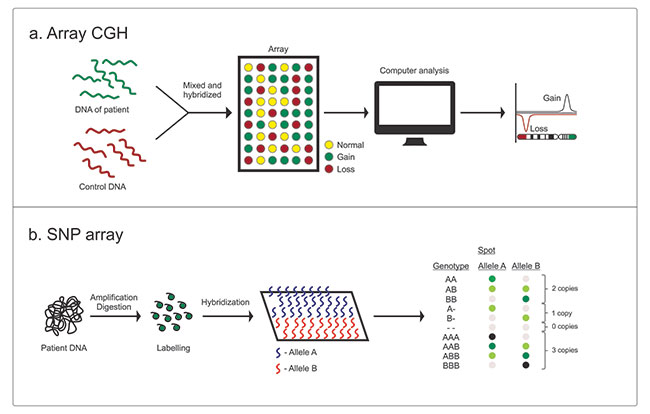 |
| Fig. 1 (a)
Comparative genomic hybridization array, and (b) Single
nucleotide polymorphism array. |
Interpretation
The variants identified are critically evaluated based on
their size, gene content and published reports in literature
[9,10]. Penetrance (how many of individuals with this variant
have a phenotypic effect) and variable expressivity (varying
severity of disease in individuals with a particular genotype)
are considered. The databases used for CNV interpretation are
given in Web Table I.
The CNVs are classified into pathogenic, benign or variant of
uncertain significance (VOUS) based on American College of
Medical Genetics and Genomics (ACMG) criteria given in
Table I. VOUS are variants, which are not directly
linked to the patient's phenotype but have some evidence for
causation. Usually laboratories using SNP arrays report variants
above 50 to 100kb in size [11]. Testing of parents may be
required to ascertain the significance of the variant.
Table I Classification of Copy Number Variants (CNVs) Based on American College of Medical
Genetics and Genomics criteria [9]
|
Type of CNVs |
|
Criteria |
|
Pathogenic |
|
• |
CNVs associated with a known microdeletion/duplication syndrome |
|
• |
CNVs reported as clinically significant in peer-reviewed journals and public databases |
|
• |
CNVs that are more than 3-5Mb size and are cytogenetically visible |
|
Uncertain clinical significance |
|
Likely pathogenic |
|
• |
CNVs reported in a single case report, but with breakpoints and phenotype correlating to the patient's features |
|
• |
CNV interval has a gene whose function is relevant to the clinical features of the patient |
|
No sub-classification |
|
• |
CNVs described in multiple peer-reviewed journals with no conclusive evidence regarding clinical significance. |
|
• |
CNV interval has genes but it is not known whether the genes are dosage sensitive |
|
Likely benign |
|
• |
CNVs are seen in small number of people in databases of variations in normal individuals |
|
• |
No gene in the CNV interval; but it is included because of the size cut off set by the laboratory |
|
Benign |
|
• |
CNVs reported as benign variants in multiple peer- reviewed publications or curated databases |
|
• |
CNVs whose benign nature has been characterized |
|
• |
CNVs represents a common polymorphism and has a population frequency of more than 1% |
CMA has the highest diagnostic yield
for any single test in evaluating cognitive impairment,
developmental delay, multiple malformations of unknown etiology
or autistic spectrum disorder [2,12]. It is the first line
investigation for antenatally detected structural abnormalities,
stillbirth or intrauterine demise [13], and when a karyotype
shows a marker chromosome or extra chromosome material of
unknown origin. CMA can identify gain or loss of chromosomal
material in up to 20% of individuals with an apparently balanced
chromosome translocation [14,15]. Box I enumerates
the advantages and disadvantages of CMA as compared to
karyotyping.
|
Box I Advantages and Limitations of Chromosomal
Microarray over Karyotyping |
|
Advantages
• CMA can be
done from DNA isolated from any type of tissue unlike
karyotyping which requires live, actively dividing
cells.
• Higher resolution: CMA detects CNVs as
small as 10 to 20 kb [9], unlike karyotype for which the
resolution is 5 Mb.
• Objective result interpretation
• Can detect cryptic imbalances in chromosomes in
apparently balanced karyotype.
Limitations
• Does
not detect balanced translocations that do not alter the
CNVs.
• Inability to detect point mutations,
deletions or duplications at the single gene level.
•
Does not detect low-level mosaicism and polyploidy.
• Missing of variations in regions that are not targeted
by the probes in targeted arrays.
• Difficulty
interpretation of VOUS.
CNV : Copy number variant;
VOUS: Variants of unknown significance.
|
One should know the design and
resolution of the testing platform and the genomic regions
covered. Most of the commercial platforms available have probes
for known microdeletion/ duplication syndromes along with genome
wide probes for other clinically significant CNVs. In a clinical
setting, a low-resolution array, covering all well-delineated
microdeletion and microduplication syndromes is usually
sufficient. High-resolution arrays are more accurate in
delineation of CNVs and SNPs, but result in a large number of
variants, which are difficult to interpret. Its utility is
limited to the research context.
Both pretest counseling (for the yield, specific benefits
and limitations) and post-test counseling are also essential.
NEXT-GENERATION SEQUENCING
NGS, also known as massively parallel sequencing or deep
sequencing, is a high throughput sequencing technology which
allows simultaneous sequencing of millions of DNA base pairs at
a comparatively lower cost and higher speed. Exomes comprise
only 1% of 6.2 billion base pairs in human DNA, which code for
proteins [16]. NGS can analyze the whole genome (whole genomic
sequencing, WGS), exome (exome sequencing, ES) or a targeted
region of interest in the human genome (targeted gene panel
testing). The features of WGS, ES and targeted sequencing are
summarized in Table II.
The steps involved are illustrated in
Web Fig. I. Depth
of sequencing is the number of times a nucleotide is read during
sequencing. A depth of 20x implies that a particular variant or
nucleotide is sequenced 20 times. Coverage usually refers to the
fraction of the target region of interest sequenced
satisfactorily (usually at least 20 times or 20x).
| Table II Characteristics of
NGS Based Tests |
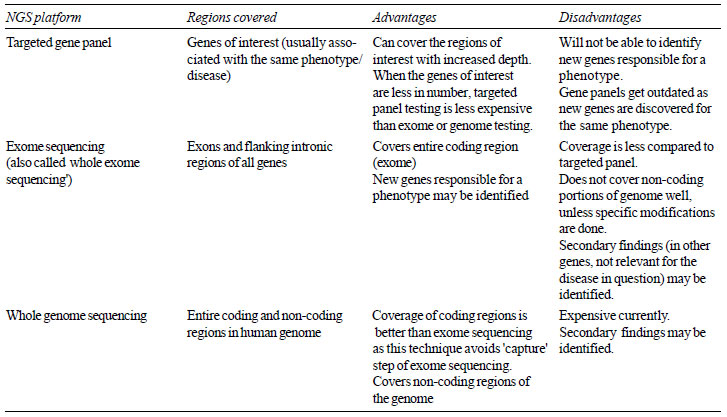 |
Interpretation
The variants are sorted to narrow down to a single variant
that is likely to explain the disease or phenotype. As monogenic
diseases are rare, it is assumed that the disease-causing
variant is usually not seen in genomes of healthy individuals in
the population. Disease-causing variants are likely to result in
a change in quantity or quality of the protein coded by the
gene, thus affecting the function of the protein. They are also
likely to be conserved across different species. Several
computational tools are now available to predict the effect of a
change in the nucleotide sequence of a gene. The sorting (also
popularly called filtering) is also aided by published databases
of normal variants and disease-causing variants (Web
Table II). If in-house databases with frequency of
variants in a particular population are available, they can be
very powerful tools for variant analysis as we expect unique
genetic variations in different ethnicities. In 2015, ACMG
published guidelines for interpretation of sequence variants and
categorized them into five categories, i.e., pathogenic, likely
pathogenic, benign, likely benign and VOUS [17]. The results are
then correlated with clinical features and communicated to the
patient. For efficient filtering and clinical interpretation of
the variants, a patient should be referred to a trained clinical
geneticist.
NGS testing generates a large number of variants in an
individual's exome or genome. Clues from evaluation of pedigree,
clinical examination and routine medical tests are vital to
determine the effect of the variant on the phenotype. Often
Human Phenotype Ontology [HPO] terms are used for this purpose.
NGS should not be considered as an alternative for thorough
clinical examination and ancillary laboratory tests.
Clinical Indications
•
Targeted panel testing can be done when a particular
phenotype is caused by variations in more than one gene (locus
heterogeneity). For example, variations in about 20 different
genes are implicated in osteogenesis imperfecta. A panel, which
covers all the genes for osteogenesis imperfecta is more
efficient than Sanger sequencing one gene after the other. Other
examples are deafness, Noonan syndrome (RASopathies), congenital
myopathy and pediatric epilepsy. Large genes like dystrophin can
be tested by NGS either singly or in a panel for muscular
dystrophy or myopathy when deletion and duplications are ruled
out by multiplex ligation dependent probe amplification (MLPA)
in a child with Duchenne muscular dystrophy.
•
ES can be performed in patients with genetically
heterogenous monogenic disorders when targeted panel testing
fails.
•
WGS may be considered when ES fails to identify a
disease-causing variant. It detects variants in coding and
non-coding regions of the genome and regions not well captured
and sequenced in ES, CNVs and structural chromosomal
abnormalities. It has the potential to become a single test
replacing most of the current tests.
•
NGS-based tests hold promise in area of carrier testing,
pre-symptomatic testing, pharmacogenetic testing, and predictive
testing, which are beyond the scope of this review.
Even though genome sequencing and exome sequencing are described
as 'whole' genome or 'whole' exome sequencing, they do not
evaluate all the genes in the human genome. The word 'whole'
distinguishes these tests from panel testing and should not
mislead clinicians and patients to believe that these tests
would be 100% sensitive to detect all the disease-causing
variants. The coverage of known genes by these tests vary from
85%-92% [18]. ‘Clinical exome’ or ‘focused exome’ is a
commercial panel test that uses a customized capture kit to
interrogate only genes associated with a known clinical
phenotype, usually listed in Online Mendelian Inheritance in Man
(OMIM). Hence the term 'clinical exome' is better avoided. In
strict sense, 'clinical' genome or exome sequencing implies
sequencing of exome or genome for clinical applications [19].
Before ordering a test, it is essential to check the coverage of
genes of interest. The decision whether to order a targeted
panel test or ES or WGS will depend on the clinical features of
a patient and the ability of a clinician to arrive at a
diagnosis. An ideal
targeted panel test should be able to diagnose disease-causing
variants in the genes of interest of the suspected genetic
disorder and should also include methods to detect deletion and
duplications, which can cause a specific disease phenotype.
Analyzing only selected regions or genes of interest may not
qualify to be called a targeted panel, unless the laboratory
fills the gaps in sequencing by alternate methods like Sanger
sequencing and does a deletion/ duplication analysis. For
example, in a child with leukodystrophy, before ordering a
targeted panel test for leukodystrophy, it is essential to check
whether all the genes of interest are covered. Krabbe disease is
often caused by deletions in GALC gene and might be missed if an
NGS test is ordered without asking for deletion/duplication
analysis of GALC gene. If a specific genetic diagnosis cannot be
made, ES or WGS may be considered. ES is cheaper and is often
preferred to WGS as the first investigation for undiagnosed
single gene diseases, which mostly result from variations in
exons. A singleton or single exome means exome sequencing of a
proband, whereas 'trio' exome means exome sequencing of the
proband and parents.
Consent and Counseling in NGS Tests
Informed consent is
essential before NGS based testing. Pretest counseling is
essential to explain the yield, utility and implications of a
‘negative’ or ‘positive’ report for family. Limitations of
science in interpreting VOUS and identification of secondary
variants are specific issues in NGS testing.
Secondary variants in genes are associated with diseases
unrelated to the proband’s condition and are common in ES and
WGS. Secondary findings in genes causing cancer and sudden
cardiac death may have implications for the patient and family
members. A genetic
diagnosis may not have any direct impact on the treatment of the
patient but may aid in long-term management, genetic counseling
and prenatal diagnosis. Post-test counseling by a geneticist is
thus needed. Sanger sequencing is done to validate the variant
in the proband and for segregation analysis. Good quality NGS
often obviates the need for Sanger confirmation. Segregation
analysis determines segregation of the variants in the other
affected or unaffected members in the family and is crucial for
causal association in the proband. If a negative test result is
obtained, the family should be counseled about the need to
re-evaluate the data at a later date.
At present there are no regulations
governing clinicians, laboratories and counselors in India.
Direct marketing of these tests may result unregulated
commercialization.
Variables to
Consider in NGS Report
The NGS report
mentions the methodology, capture kit, depth and coverage of
sequencing. Capture kits may be customized for different panel
tests and ES. It is
important to check for depth and coverage of sequencing before
conveying the report to the patient.
Some clinical scenarios where CMA
and NGS have aided in diagnosis are described in
Web Table III.
CONCLUSIONS
Chromosomal
microarray, exome sequencing and whole genome sequencing using
NGS techniques are powerful methods to investigate variations in
human genome. It is essential for a pediatrician to know the
strengths, limitations and advantages of these testing methods
over traditional medical tests to apply optimally in clinical
practice of pediatrics.
Contributors:
DLN: substantial contributions to design and draft of the work;
GKM: substantial contributions to the conception and design of
the work, drafting and revising it critically for important
intellectual content. Both approve the final version to be
published and agree to be accountable for all aspects of the
work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Funding:
None; Competing interest: None stated.
REFERENCES
1. Bernardini L, Alesi V,
Loddo S, Novelli A, Bottillo I, Battaglia A, et al.
High-resolution SNP arrays in mental retardation diagnostics:
How much do we gain? Eur J Hum Genet. 2010;18:178-85.
2. Manning M, Hudgins L.
Array-based technology and recommendations for utilization in
medical genetics practice for detection of chromosomal
abnormalities. Genet Med. 2010;12:742-5.
3. Miller DT, Adam MP,
Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al.
Consensus Statement: Chromosomal Microarray is a First-tier
Clinical diagnostic Test for Individuals With Developmental
Disabilities or Congenital Aanomalies. Am J Hum Genet. 2010;86:
749-64.
4. Snijders AM, Nowak N,
Segraves R, Blackwood S, Brown N, Conroy J, et al.
Assembly of microarrays for genome-wide measurement of DNA copy
number. Nat Genet. 2001;29:263-4.
5. Beaudet AL. The utility
of chromosomal microarray analysis in developmental and
behavioral pediatrics. Child Dev. 2013;84:121-32.
6. Oostlander AE, Meijer
GA, Ylstra B. Microarray-based comparative genomic hybridization
and its applications in human genetics. Clin Genet.
2004;66:488-95.
7. LaFramboise T. Single
nucleotide polymorphism arrays: A decade of biological,
computational and technological advances. Nucleic Acids Res.
2009;37:4181-93.
8. Feuk L, Carson AR,
Scherer SW. Structural variation in the human genome. Nat Rev
Genet. 2006;7:85-97.
9. Kearney HM, Thorland EC,
Brown KK, Quintero-Rivera F, South ST. American College of
Medical Genetics Standards and Guidelines for Interpretation and
Reporting of Postnatal Constitutional Copy Number Variants.
Genet Med. 2011;13:680-5.
10. South ST, Lee C, Lamb
AN, Higgins AW, Kearney HM. ACMG Standards and Guidelines for
Constitutional Cytogenomic Microarray Analysis, Including
Postnatal and Prenatal applications: Revision 2013. Genet Med.
2013;15:901-9.
11. Levy B, Wapner R.
Prenatal diagnosis by chromosomal microarray analysis. Fertil
Steril. 2018;109:201-12.
12. Rauch A, Hoyer J, Guth
S, Zweier C, Kraus C, Becker C, et al. Diagnostic yield
of various genetic approaches in patients with unexplained
developmental delay or mental retardation. Am J Med Genet A.
2006;140:2063-74.
13. Karampetsou E, Morrogh
D, Chitty L. Microarray technology for the diagnosis of fetal
chromosomal aberrations: Which platform should we use? J Clin
Med. 2014;3:663-78.
14. Edelmann L, Hirschhorn
K. Clinical utility of array CGH for the detection of
chromosomal imbalances associated with mental retardation and
multiple congenital anomalies. Ann NY Acad Sci.
2009;1151:157-66.
15. Sismani C,
Kitsiou-Tzeli S, Ioannides M, Christodoulou C, Anastasiadou V,
Stylianidou G, et al. Cryptic genomic imbalances in
patients with de novo or familial apparently balanced
translocations and abnormal phenotype. Mol Cytogenet. 2008;1:15.
16. Thiffault I, Lantos J.
The challenge of analyzing the results of next-generation
sequencing in children. Pediatrics. 2016;137:S3-7.
17. Richards S, Aziz N,
Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards
and Guidelines for the Interpretation of Sequence Variants: A
Joint Consensus Recommendation of the American College of
Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 2015;17:405-24.
18. Biesecker LG, Biesecker
BB. An approach to pediatric exome and genome sequencing. Curr
Opin Pediatr. 2014;26:639-45.
19. Biesecker LG, Green RC.
Diagnostic clinical genome and exome sequencing. N Engl J Med.
2014;371:1170.
20. Schwarze K, Buchanan J,
Taylor JC, Wordsworth S. Are whole-exome and whole-genome
sequencing approaches cost-effective? A systematic review of the
literature. Genet Med. 2018;20:1122-30.
21.Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, et al. Clinical whole-exome sequencing
for the diagnosis of mendelian disorders. N Engl J Med.
2013;369:1502-11.


