History and examination: A
3-year-old boy was admitted with fever and progressive pallor of 2
months duration. He was asymptomatic till 2 months ago, when he started
developing high grade fever with intermittent spikes once every day. He
would be relatively well in between two fever spikes. Fever was
associated with progressive pallor requiring one blood transfusion 20
days back. He had documented thrombocytosis (platelet count: 7 lakhs/mm
3)
at that time. There was no history of localizing features for fever.
Past history, developmental history and immunization history were not
contributory. There was history of one sibling death at birth for which
there was no obvious cause. Examination revealed significant pallor.
There was absence of icterus, edema, clubbing, cyanosis, lymphadenopathy,
and rash. He was febrile at admission. He weighed 9.5 Kg (expected 14
kg), and had a height of 84 cm (expected 92 cm) and occipitofrontal
circumference (OFC) of 46 cm. Abdominal examination showed
hepatosplenomegaly with liver of 3 cm below costal margin and spleen of
2 cm below costal margin. Chest, cardiovascular, neurological and joint
examinations were normal.
TABLE I Serial Hematological and Biochemical Parameters of the Index Patient During the Hospital Stay
|
Day of hospital stay |
Day 2 |
Day 7 |
Day 16 |
Day 22 |
Day 28 |
|
Total leucocyte counts (cells/mm3) |
17,600 |
8,500 |
4,500 |
16,000 |
300 |
|
Differential counts (P/L/M/E)# |
59/31/10 |
06/77/16/01 |
48/31/20/01 |
26/56/15/01 |
Too low |
|
Hemoglobin (g/dL) |
8.1 |
6.0 |
9.3 |
9.9 |
5.9 |
|
Platelets (cells/mm3) |
1.95 lakhs |
1.27 lakhs |
4.82 lakhs |
13,000 |
1000 |
|
ESR* |
62 |
- |
59 |
- |
02 |
|
CRP* (mg%) |
254 |
234 |
248 |
436 |
279 |
|
Blood Urea/ Creatinine (mg/dL) |
15/ 0.5 |
17/ 0.5 |
18/ 0.5 |
214/ 2.0 |
193/ 2.7 |
|
ALT/ AST* (U/L) |
155/ 26 |
76/ 25 |
190/ 37 |
482/ 63 |
53/ 33 |
|
Total proteins/ albumin (g/dL) |
7.9/ 2.7 |
6.8/ 2.1 |
7.0/ 2.2 |
5.7/ 1.9 |
3.9/ 2.3 |
|
Serum ferritin (ng/mL) |
- |
44,638 |
16,500 |
- |
5258 |
|
Fasting triglycerides (mg/dL) |
93 |
441 |
- |
189 |
- |
|
PT/ APTT* (sec) |
19/ 28 |
18/ 25 |
16/ 25 |
21/ 39 |
19/ 38 |
|
Fibrinogen (g/L) |
4.83 |
3.9 |
- |
0.60 |
2.18 |
|
Urine albumin$ |
1+ |
– |
– |
– |
2+ |
|
Urine microscopy |
WBC casts+ |
– |
– |
– |
– |
|
RBCs/hpf |
– |
– |
– |
– |
10-15 |
|
# Differential counts in order of polymorphs,
lymphocytes, monocytes and eosinophils; *ESR: Erythrocyte
Sedimentation Rate, CRP: C Reactive Protein, ALT: Alanine
Transaminase, AST: Aspartate Transaminase, PT: Prothrombin time,
APTT: Activated Partial Thromboplastin time; $Urine albumin by
dipstick assay. |
Investigations: Investigations revealed
anemia and polymorphonuclear leucocytosis with normal platelet counts (Table
I). Peripheral blood film showed microcytic hypochromic erythrocytes
with hypersegmented polymorphs and there were no features of hemolysis.
Erythrocyte sedimentation rate (ESR) was raised and he had
hypoalbuminemia and high aspartate transaminase levels. Extensive
infection workup (Box I) was negative except for a
positive serology for Epstein Barr virus (EBV) viral capsid antigen
(VCA) IgM. Fine needle aspiration cytology (FNAC) from axillary lymph
node was suggestive of reactive lymphoid hyperplasia. Bone marrow had no
evidence of leukemia or hemophagocytosis. Chest X-ray (CXR) was
normal and ultrasound abdomen showed hepatosplenomegaly. Contrast
enhanced computerized tomography (CECT) of chest and abdomen showed
hepatosplenomegaly, bilateral pleural effusion and underlying lung
collapse. Immunological work up was normal except for low C4 levels (Box
I).
|
Box I Investigations for Prolonged Pyrexia in the Index
Patient |
|
Blood cultures: Sterile on day 1, 5, 7, 16, 21
Urine cultures: Sterile on
day 1, 5, 22
Blood fungal cultures:
Sterile
Fungal serology: Negative
Gastric lavage for AFB: No
AFB seen
Brucella/HIV/ Parvovirus
serology: Negative
IgM scrub typhus: Negative
EBV serology (VCA IgM):
Positive
Pericardial fluid analysis:
No cells, no organism on Gram stain, Bacterial and fungal
cultures sterile, AFB smear and cultures sterile
Investigations for malignancy
FNAC axillary node: Cellular;
Reactive population of lymphoid cells with scattered histiocytes
and tingible body macrophages. There were no atypical cells or
hemophagocytosis. Stain for AFB was negative
Bone marrow examination:
Smears were cellular with myeloid to erythroid ratio of 3.4:1.
Erythropoiesis was normoblastic and megakaryocytes were adequate
on smears. Differential counts in marrow were normal and blasts
were 2%. There was no significant hemophagocytosis. No
microorganisms were noted. Trephine biopsy was normal and there
was no granuloma and lymphoid aggregates.
Immunological work-up
Antinuclear antibody by
indirect immunofluorescence: Negative
Anti-neutrophil cytoplasmic
antibody (ANCA): Negative
Perforin expression:
Normal by flow cytometry
Nitroblue Tetrazolium
test (NBT): Normal reduction
C3, C4*: 162 mg (50-150
mg), 4 mg (20-50 mg)
Lymphocyte subsets*:
CD3- 71.7% (43-76%), CD19- 14.9% (14-44%), CD56- 9.5% (4-23%)
*Numbers in parenthesis indicate the normal ranges for the
age.
|
Course and management: Possibility of
infections and malignancy was kept at admission. The index child was
started on intravenous Ceftriaxone, Cloxacillin and oral Doxycycline. He
developed increasing lymph nodes, worsening respiratory distress and
lethargy with progressive neutropenia during hospital stay. Chest X-ray
done during second week of hospital stay showed patchy areas of collapse
and consolidation. Antibiotics were changed to injectable Vancomycin and
Imipenem. In view of very high serum ferritin, a possibility of
Hemophagocytic lymphohistiocytosis (HLH)/ Macro-phage Activation
Syndrome (MAS) was kept and he was given pulse intravenous
Methylprednisolone 30 mg/kg/day for five days. There was a brisk
response in fever and respiratory distress. Size of liver, spleen and
lymph nodes decreased. He was subsequently shifted to oral prednisolone
2 mg/kg/day. He started developing fever again on day 2 of oral
steroids. He was restarted on IV Methylprednisolone and as he did not
show a significant response, he was given a dose of intravenous
immunoglobulin (IVIg). He responded in terms of reduction in fever and
respiratory distress. He developed sudden onset severe respiratory
distress and shock on day 20 of admission. Chest X-ray showed
enlarged globular heart and echocardiography revealed significant
multiloculated pericardial effusion causing tamponade. Emergency
pleuropericardial window was created under general anaesthesia.
Intra-operative findings were thickened pericardium with multiple
loculations and 200 ml of seropurulent fluid was drained. Pericardial
tissue histopathology was suggestive of fibrinous pericarditis.
Vasopressors, Fluconazole and Clindamycin were added. However, he kept
on worsening with development of oliguric renal failure requiring three
cycles of peritoneal dialysis. He also needed multiple packed red cell
and platelet transfusions. He succumbed to refractory shock with
multiorgan dysfunction on day 30 of hospital stay. Postmortem
cerebrospinal fluid examination was normal.
Unit’s final diagnosis: Hemophagocytic
lympho-histiocytosis (HLH)/ Macrophage Activation Syndrome (MAS),
Systemic JIA (SJIA)
Discussion (Clinical discussant): Diagnosis
of HLH is not in doubt in the index child as he did satisfy five
clinical criteria for HLH: fever, splenomegaly, cytopenia involving two
cell lines, high fasting triglycerides and high serum ferritin [1]. He
also showed brisk response to steroids in terms of reduction in fever,
respiratory distress and increase in platelet counts. HLH is a syndromic
diagnosis and no single clinical finding or lab test is diagnostic of
this condition [1]. However, ferritin levels more than 10,000 ng/ml have
been found to be 98% specific for HLH in children [2]. Absence of
hemophagocytosis in various tissues does not exclude HLH [3].
There are various genetic and acquired factors, which
can cause HLH and frequently one can get more than one underlying cause
[1]. Usually, HLH occurs in a genetically predisposed individual when
one or more acquired factors like infections, malignancies and/or
rheumatological diseases act as trigger. Presence of EBV VCA IgM
positivity suggests acute EBV infection, which is the most common
acquired cause known to precipitate HLH [4]. The parameters odd for EBV
HLH in the index child are prolonged duration of fever,
polymorphonuclear leucocytosis, thrombocytosis, very high ESR and
fibrinogen at admission. Hence, to explain prolonged features of
inflammation prior to presentation as HLH, I would like to discuss other
underlying causes.
Coming to rheumatological conditions, SJIA is a
strong possibility as the index child had typical fever pattern,
serositis, hepatosplenomegaly, polymorpho-nuclear leucocytosis and
thrombocytosis at the onset. Absence of arthritis does not exclude SJIA.
HLH/MAS can be seen at first presentation in SJIA [5] and prolonged
preceding inflammation suggests a diagnosis of SJIA. So, we have
evidence of HLH in this child with underlying SJIA and acute EBV
infection being a trigger.
Is it possible that this child had a genetic
predisposition to develop HLH? Genetic causes of HLH include familial
HLH, HLH associated with immunodeficiency syndrome with albinism,
X-linked lymphoproliferative disorder (XLP) and X-linked inhibitor of
apoptosis deficiency. Apart from younger age at presentation,
differentiation between genetic and acquired causes based on clinical
presentation alone is difficult [6]. XLP is associated with EBV-related
HLH, lymphoma and hypogammaglobulinemia. HLH in XLP cannot be
differentiated from HLH of secondary etiologies [7]. A considerable
proportion of EBV HLH patients have been shown to have underlying
genetic defects [8]. It is impossible to rule out underlying genetic
causes with the available investigations.
During third week, this child had recurrence of
fever, respiratory distress and pericardial effusion, low ESR, high CRP,
and transaminitis. He went on to develop pancytopenia, renal
dysfunction, shock, coagulopathy and low fibrinogen. Fibrinous
pericarditis as seen in this child has been described in a few cases of
HLH [9]. Similarly, renal involvement here could be multifactorial,
related to shock as well as to cytokine nephropathy [10].
This child also had unexplained low C4. Congenital C4
deficiencies are associated with autoimmune diseases [11]. SJIA, in
contrast, is an autoinflammatory condition and it is not commonly known
to be associated with low C4. Terminal events seem to be worsening of
MAS with multiorgan dysfunction syndrome (MODS). However, it is
difficult to exclude a diagnosis of nosocomial sepsis. So HLH/MAS with
MODS with an underlying SJIA, with trigger being an acute EBV infection
seems likely in this child.
Open Forum
Pediatrician 1: Diagnosis of SJIA/MAS does not
seem to be in doubt in the index case. Although arthritis is required
for the diagnosis of SJIA by the classification criteria, there is a
subset of patients who develop arthritis years after onset of fever.
Preterminally, we can see falling ferritin and there is progressive
cytopenia. So, there is a high chance that this child would have
acquired nosocomial infection, especially fungal.
Pediatric Hematologist: The context here seems to
be what is the cause of HLH, whether it is familial or SJIA. Histology
cannot pinpoint the etiology of HLH. Steroids when used alone for
treating HLH can lead to transient response and without Etoposide, there
can be a recurrence (12).
Pediatrician 2: In a child with prolonged fever,
thrombocytosis and hyperferritinemia with values in several thousands,
the differential diagnosis narrows down to SJIA. The trouble in the
index child is that this child presented with a complication of SJIA
right at the onset. So, as I look at it, this child had SJIA with MAS
and he succumbed to MAS.
Adult physician 1: How frequent is JIA under age
of five? Do we believe that seropurulent pericardial effusion with 200
ml of fluid was related to SJIA/ HLH?
Pediatrician 3: There is no doubt about diagnosis
of SJIA. In this patient, once Methylprednisolone worked and after that
whatever happened, looks like a secondary infection.
Clinical discussant: SJIA is predominantly a
disease of under-fives. Arthritis, if present, would have made diagnosis
of SJIA easier. MAS can be the initial presentation of SJIA. Infections
are associated with an increase in ESR and CRP. The index child, had a
raised CRP but the ESR was very low. This could explain MAS as the
predominant cause for preterminal worsening. Nosocomial infections
continue to be another possibility for preterminal worsening and are a
major cause of mortality in HLH/MAS.
Adult physician 2: I do not believe that HLH got
any better in this child. There was a progressive fall in counts and
what happened after steroids and IVIg, was just a mild decrease in
inflammation.
Pathology Protocol
Antemortem pericardial biopsy showed fibrin rich
exudates with paucity of inflammatory cells and was reported as
fibrinous pericarditis. There were no granulomas or malignancy in the
pericardial biopsy.
This was a partial autopsy and the prosector noted
gangrene of right little finger and right toe, rashes over abdomen and
anal excoriation. No excess fluid was seen in the serous cavities.
Discolored and hemorrhagic mucosa and serosal exudates were seen in
ileum, ileocecal region and large intestine. Ulcerations of variable
sizes (0.5 to 5 cm diameter) with hemorrhagic base and dirty exudates
were seen involving the entire caecum and terminal part of the small
intestine. Microscopically, small intestine showed extensive hemorrhagic
ulceration of the entire mucosa with involvement of muscle causing
myocytolysis and no preserved epithelium was seen. Many fungal profiles
and thrombi were seen in blood vessels in the intestinal wall. There was
hardly any inflammatory reaction. In addition, colonies of bacteria were
seen in the small and large intestine, both on serosal and mucosal
aspects. The esophagus and stomach showed large kissing ulcerations.
Some of them appeared hemorrhagic with greenish exudates in base and
sharp outline (Fig. 1a). Fig 1b highlights
the septate fungal profiles infiltrating into gastric mucosa causing
ischemic necrosis. Periodic acid–Schiff (PAS) and Groccot’s stain showed
septate fungal profiles with acute angle branching. There was absolute
paucity of inflammatory cells in the ulcerated area. The section from
appendix showed preserved lymphoid follicles.
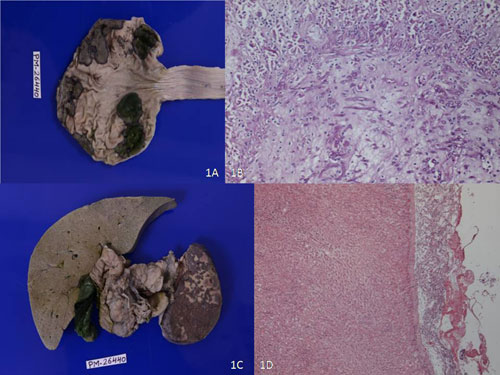 |
|
Fig. 1 (a) Gross photograph
showing punched out bile stained ulcers in the stomach; (b)
Microphotograph showing ulcerated mucosa with septate fungal
profiles in stomach (H&Ex20X); (c) Gross photograph showing bile
stained liver and splenic infarct; (d) Microphotograph showing
septate fungal profiles over capsular surface of liver
infiltrating parenchyma (H&Ex10X).
|
Surface of the liver showed multiple superficial
nodular whitish lesions of 0.5 to 5 cm diameter. Microscopic examination
revealed fibrinous exudate with sheets of fungal profiles extending into
the parenchyma, causing infarction (Fig. 1d). Fungal
profiles were also identified in the portal vein. Vessels surrounding
the mesenteric perinodal tissue and lymph node sinusoids showed fungal
thrombi.
Liver was grossly enlarged with exaggerated mottling.
Microscopic examination of the liver showed diffuse micro- and
macro-vesicular steatosis, sinusoidal dilatation and congestion.
Reticulin framework was maintained and there was no fibrosis in the
portal tract. Further magnification showed focal cholestasis in liver
and prominence of Kupffer cells. There was no hemophagocytosis in liver.
Spleen was enlarged (weight 80 grams) and showed
multiple geographic infarcts (Fig. 1c). Infarcts with
hemorrhagic borders were confirmed on microscopy. There was evidence of
perisplenitis. The preserved white pulp could be seen around the splenic
arterioles and surrounding parenchyma showed macrophage proliferation.
In addition, lot of nuclear debris was seen around the arterioles.
However, no evidence of hemophagocytosis was noted.
Lymph nodes showed presence of lymphoid tissue,
benign sinus hisitiocytosis, expansion of sinuses and congested vessels.
However, no evidence of hemophagocytosis was noted here too. There was
relative depletion of B cells in some of the follicles; however, T cells
were preserved.
Lungs weighed 180 grams and were subcrepitant to
feel. There was patchy pleural thickening with subpleural hemorrhages (Fig.
2a). Thymus could not be delineated. Tracheobronchial tree showed
diffuse ulcerations with greenish brown dirty exudates. There was no
evidence of pulmonary thrombus. Microscopic sections showed fibrinous
pleuritis with proliferation of macrophages in the pleura confirmed by
CD68 stain. Other parts of lungs showed exudation of macrophages within
alveolar spaces with colonies of bacteria occupying almost all alveoli (Fig.
2 b). PAS stain showed faint positivity within the bacterial
colonies. Gram’s stain identified them as red indicating gram negative
colonies.
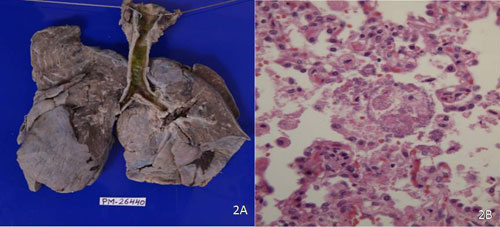 |
|
Fig. 2 (a) Gross photograph of both
lungs with consolidation and greenish exudate in
tracheobronchial tree; (b) Microphotograph showing bacterial
colonies within the alveoli without inflammation (H&Ex20X). (See
color image at website).
|
Kidneys were swollen with flea bitten appearance and
pin point hemorrhages and weighed 130 grams. The cut surface showed
microinfarcts. In addition, there were fibrin thrombi in the glomerular
capillary loops and there was no evidence of glomerulonephritis.
The heart weighed 70 grams and showed features of
fibrinous pericarditis. The endocardium appeared opaque on left side
inflow and outflow portion with dilatation. Microscopically there was
evidence of pericarditis with scattered inflammatory cells and
macrophage proliferation. Sections from myocardium showed subendocardial
and parenchymal calcifications. One section from the heart revealed
fungal thrombi within the veins as well as arteries of the pericardium.
Biopsy of the skin showed intact epidermis and
infracted adnexal structures. There were congested vessels with fibrin
thrombi within them, indicating it to be a part of disseminated
intravascular coagulation (DIC). Testes also showed fibrinoid necrosis
and fibrin thrombi within vessel lumen.
The final autopsy diagnosis in this 3-year-old boy
diagnosed as SJIA is
• Primary gastrointestinal aspergillosis with
serosal spread and dissemination to lymph nodes, mesentery, liver
and pericardial vessels
• Splenic infarct secondary to thrombosis
• Evidence of DIC in skin, kidneys and testes
• Gram negative lobar pneumonia with
tracheobronchial ulceration, pericarditis and pleuritis.
Open Forum
Adult physician 1: Thrombocytopenia which
occurred in this patient is probably related to DIC secondary to
infection.
Pediatrician 2: The primary diagnosis would still
be SJIA/MAS with nosocomial fungal sepsis. Hemophagocytosis would have
disappeared with treatment. MAS in this setting has a very high
mortality which can be reduced by using Anakinra (13), an interleukin-1
receptor antagonist, which is currently not available in India.
Pediatrician 3: Is this kind of massive
Aspergillus enteritis common and is there any way to make an antemortem
diagnosis?
Pathologist 1: Antemortem diagnosis can be made
with help of galactomannan and fungal PCRs, but there are issues of
false positivity in them too.
Adult physician 2: We see lot of histiocytes in
the pericardium and lymph nodes. Is it not usual for a fungal infection
to produce a histiocytic response? What is the explanation for GI
mucosal infarcts and myocardial calcification?
Chairman: There are some questions which are yet
to be answered. But in conclusion, it appears that the index child had
SJIA to start with and later on succumbed to multiple secondary
infections.
Discussion
The clinical course in the index child reiterates the
fact that HLH is a syndromic diagnosis and all diagnostic criteria may
not be present initially [14], delineating the importance of serial
follow up. Absence of hemophagocytosis in various tissues does not
exclude a diagnosis of HLH/MAS as it is not always present at the
initial marrow examination and serial examinations may reveal its
presence [14,15]. We could not demonstrate hemophagocytosis in index
child even at autopsy. This could have been due to treatment with
steroids and IVIg. In an autopsy series of 27 children with HLH, three
did not have hemophagocytosis in post-mortem histopathology because of
treatment with immunosuppressive drugs [16].
Underlying causes for HLH could be genetic or
acquired [4]. In a majority of cases, HLH occurs secondary to one or
more acquired triggers in a setting of genetic predisposition [4, 17].
EBV is said to be the most common acquired trigger as was seen in a
Japanese registry where more than 40% cases of childhood HLH were
related to EBV infection [18]. Mean age at presentation in this registry
was 3.9 years [18]. Genetic mutations have been identified in a
significant proportion of patients with HLH who have an identifiable
acquired trigger [17].
MAS is a term used to describe hemophagocytosis in
association with a rheumatological disorder [19]. In a study done to
compare features of 27 SJIA/MAS patients with 90 familial HLH and 42
virus-associated HLH patients, absolute neutrophil count > 1800 cells/mm3
at onset and CRP >90 mg/L were found to indicate MAS/SJIA [6]. Once
recognized, early aggressive therapy for MAS is essential as mortality
in SJIA/MAS is reported to be around 8% in a multicentric study of 362
patients [20]. High dose corticosteroids, cyclosporine, anakinra and
IVIg constitute front-tier therapeutic choices in SJIA/MAS [21].
Independent predictors of early fatal outcome in HLH
have been found to be platelet count < 75,000 cells/mm
1. Janka G. Hemophagocytic lymphohistiocytosis: A
serious challenge for every physician. Rev Clin Esp. 2014;214: 318-9.
2. Allen CE, Yu X, Kozinetz CA, McClain KL. Highly
elevated ferritin levels and the diagnosis of hemophagocytic
lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:1227-35.
3. Bode SF, Lehmberg K, Maul-Pavicic A, Vraetz T,
Janka G, Stadt UZ, et al. Recent advances in the diagnosis and
treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther.
2012;14:213.
4. Janka G. Hemophagocytic lymphohistiocytosis: when
the immune system runs amok. Klin Padiatr. 2009;221:278-85.
5. Vastert SJ, Prakken BJ. Paediatric rheumatic
disease: Diagnosing macrophage activation syndrome in systemic JIA. Nat
Rev Rheumatol. 2014;10:640-2.
6. Lehmberg K, Pink I, Eulenburg C, Beutel K, Maul-Pavicic
A, Janka G. Differentiating macrophage activation syndrome in systemic
juvenile idiopathic arthritis from other forms of hemophagocytic
lymphohistiocytosis. J Pediatr. 2013;162:1245-51.
7. Faitelson Y, Grunebaum E. Hemophagocytic
lymphohistiocytosis and primary immune deficiency disorders. Clin
Immunol. 2014;155:118-25.
8. Liu R, Shi X, Li J, Hu T, Cao J, Sun Y, et al.
[Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in
Chinese children: a primary study on clinical characteristics and gene
mutations]. Zhonghua Yi Xue Za Zhi. 2014;94:1941-6.
9. Rigante D, De Rosa G, Bertoni B, Ansuini V, Pardeo
M, La Torraca I, et al. Large pericardial effusion requiring
pericardiocentesis as cardinal sign of macrophage activation syndrome in
systemic onset-juvenile idiopathic arthritis. Rheumatol Int.
2007;27:767-70.
10. Nagayama Y, Yoshimura A, Iwasaki S. Cytokine
nephropathy in a patient with fatal Epstein-Barr virus-associated
hemophagocytic syndrome. Ren Fail. 2013;35:1445-8.
11. Bryan AR, Wu EY. Complement deficiencies in
systemic lupus erythematosus. Curr Allergy Asthma Rep. 2014;14:448.
12. Henter JI, Arico M, Egeler RM, Elinder G, Favara
BE, Filipovich AH, et al. HLH-94: a treatment protocol for
hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte
Society. Med Pediatr Oncol. 1997;28:342-7.
13. Ravelli A, Grom AA, Behrens EM, Cron RQ.
Macrophage activation syndrome as part of systemic juvenile idiopathic
arthritis: Diagnosis, genetics, pathophysiology and treatment. Genes
Immun. 2012;13:289-98.
14. Henter JI, Horne A, Arico M, Egeler RM,
Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and
therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr
Blood Cancer. 2007;48:124-31.
15. Rosado FG, Kim AS. Hemophagocytic
lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin
Pathol. 2013;139:713-27.
16. Ost A, Nilsson-Ardnor S, Henter JI. Autopsy
findings in 27 children with haemophagocytic lymphohistiocytosis.
Histopathology. 1998;32:310-6.
17. Cetica V, Sieni E, Pende D, Danesino C, De Fusco
C, Locatelli F, et al. Genetic predisposition to hemophagocytic
lymphohistiocytosis: Report on 500 patients from the Italian registry. J
Allergy Clin Immunol. 2016;137:188-96.
18. Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H,
Miura I, et al. Nationwide survey of hemophagocytic
lympho-histiocytosis in Japan. Int J Hematol. 2007;86:58-65.
19. Janka G, zur Stadt U. Familial and acquired
hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ
Program. 2005:82-8.
20. Minoia F, Davi S, Horne A, Demirkaya E, Bovis F,
Li C, et al. Clinical features, treatment, and outcome of
macrophage activation syndrome complicating systemic juvenile idiopathic
arthritis: a multinational, multicenter study of 362 patients. Arthritis
Rheumatol. 2014;66:3160-9.
21. Schulert GS, Grom AA. Pathogenesis of macrophage
activation syndrome and potential for cytokine- directed therapies. Annu
Rev Med. 2015;66:145-59.
22. Dao AT, Luong VT, Nguyen TT, Huynh QT, Phan TT,
Lam MT, et al. Risk factors for early fatal outcomes among
children with hemophagocytic lymphohistiocytosis (HLH): a
single-institution case-series in Vietnam. Pediatr Hematol Oncol.
2014;31:271-81.
23. Trottestam H, Horne A, Arico M, Egeler RM,
Filipovich AH, Gadner H, et al. Chemoimmunotherapy for
hemophagocytic lymphohistiocytosis: long-term results of the HLH-94
treatment protocol. Blood. 2011;118:4577-84.
24. Sung L, Weitzman SS, Petric M, King SM. The role
of infections in primary hemophagocytic lympho-histiocytosis: a case
series and review of the literature. Clin Infect Dis. 2001;33:1644-8.


