|
|
|
Indian Pediatr 2013;50: 579-586
|
 |
Diagnostic Approach to Primary
Immunodeficiency Disorders
|
|
M Madkaikar, A Mishra and K Ghosh
From Department of Pediatric Immunology and
Leukocyte Biology, National Institute of Immunohaematology
(ICMR), 13th Floor, NMS Bldg, KEM Hospital, Parel, Mumbai,
India.
Correspondence to: Dr Manisha Madkaikar,
National Institute of Immunohaematology, 13th Floor, NMS Bldg,
KEM Hospital, Parel, Mumbai 400 012, India.
Email:
madkaikarmanisha@icmr.org.in
|
Primary immunodeficiency disorders (PIDs) are a heterogeneous group of
inherited disorders that affect different components of the immune
system. There are more than 150 different disorders which have been
described till date. Despite major advances in the molecular
characterization of PIDs over the last 20 years, many patients remain
undiagnosed or are diagnosed too late with severe consequences.
Recognizing different clinical manifestations of PID is the first most
important step. It should be followed by use of appropriate diagnostic
tools from a vast number of investigations available. This review will
focus on important presenting features of PID and laboratory approach
for diagnosis of suspected cases of PID.
Key words: Antibody deficiency, Phagocytic
defects, Immune dysregulation, Primary immunodeficiency disorders,
Severe combined immunodeficiency (SCID).
|
|
Primary
immunodeficiency disorders (PIDs) comprise more
than 150 different disorders that affect the
development, function, or both of the immune
system [1]. In most cases, PIDs are monogenic
disorders that follow a simple Mendelian
inheritance; however, some PIDs are of more
complex polygenic origin. All forms of PIDs are
rare and have an overall prevalence of
approximately 1:10,000 live births with the
exception of IgA deficiency. However, a much
higher rate is observed among populations with
high consanguinity or among genetically isolated
populations.
PIDs are classified into
eight major categories according to the
component of the immune system primarily
involved [1]:
1. Combined T-cell and
B-cell immunodeficiencies
2. Predominantly antibody
deficiencies
3. Other well defined
immunodeficiency syndromes
4. Diseases of immune
dysregulation
5. Congenital defects of
phagocyte number and function
6. Defects in innate
immunity
7. Autoinflammatory
disorders
8. Complement
deficiencies.
In infants and children
(early childhood), the immune system is not
fully developed and they are also exposed to
many pathogens as they mix with family members
and other children in the nursery. Therefore
recurrent infections are common in young
children. Recurrent or persistent infection is
the major manifestation of primary
immunodeficiency (PID), though the pattern,
infecting microorganisms and the severity of
infections is usually different. While most
children with recurrent infections have a normal
immune system, it is important to recognize a
child with an underlying PID from a normal child
so that further investigations can be ordered
selectively. Prompt and accurate diagnosis of
PID not only helps to direct the most
appropriate treatment, and predict prognosis,
but also it is important for further genetic
counseling for the family.
The treatment modalities for
PID mainly include immunoglobulin replacement,
antibiotics and bone marrow transplantation.
Immunoglobulin replacement and judicious use of
prophylactic antibiotics can prevent the
significant end organ damage and improve
long-term outcome and quality of life in many
patients with PID if diagnosed early [2].
Hematopoietic stem cell transplantation is used
for treating many of the severe
immunodeficiencies. In centers specialized in
treating these conditions, the survival and cure
of the disease can reach up to 95%, depending on
the condition of the patient at the time of
treatment and the donor availability [3].
Thus it is
important to recognize children with PID before
significant end organ damage occurs to maximize
the opportunity for successful treatment and a
normal lifespan.
In this review we have
highlighted the important clinical
manifestations of PIDs including the pattern of
infections which would alert the clinicians to
suspect PID and the laboratory approach required
for further evaluation of some common categories
of PID.
Recognizing Clinical
Manifestations of PID
Careful clinical evaluation
is crucial for recognition of patients with PID.
It is important to know the presenting features
and warning signs of PID in order to decide the
need for further investigations. The European
Society of Immunodeficiencies (ESID) has
suggested 10 warning signs for suspicion of PID
[4] (Box 1) [4,7].
|
Box 1 Warning Signs for Suspicion of
Primary Immuno-deficiency Disorders [4]. |
• Four or more new ear infections
within 1 year.
• Two or more serious sinus
infections within 1 year.
• Two or more months on
antibiotics with little effect.
• Two or more pneumonias within 1
year.
• Failure of an infant to gain
weight or grow normally.
• Recurrent, deep skin or organ
abscesses.
• Persistent thrush in mouth or
fungal infection on skin.
• Need for intravenous antibiotics
to clear infections.
• Two or more deep-seated
infections including septicemia.
• A family history of PID. |
Although this does not
include comprehensive list of all signs and
symptoms of PID, patients showing these signs
must be evaluated further for an underlying PID.
While evaluating such children, important
clinical features like age at presentation,
pattern of infection, non-infectious
manifestations and family history should also be
taken into consideration as these give an
important clue to the underlying immune defect
[5]. Though it is difficult to predict a
specific PID on the basis of infections with
particular organisms, they definitely provide an
important clue to underlying immune defect (Table
I). The important distinct clinical
manifestations of different categories of PID
are discussed below.
TABLE I Clues to the Presence of Primary Immunodeficiency
|
PID Category |
Infectious complications |
Organisms |
Diagnostic tests |
|
Combined T and |
Systemic viral |
Bacteria: Pyogenic bacteria |
T cells: |
|
B cell deficiency |
infections, |
Campylobacter Listeria |
Lymphocyte subsets: |
|
|
gastroenteritis |
|
1. T, Tc, Th, B , NK |
|
|
|
|
2. DNT cells |
|
|
|
|
3. Memory, naïve and activated T cell |
|
|
|
Viruses: All, especially, respiratory
|
Specific cell surface antigen
expressions: |
|
|
|
syncytial virus, EBV, parainfluenza
|
1. CD132: |
|
|
|
type 3 |
2. CD127 |
|
|
|
Fungi:Candida, Aspergillus |
3. CD154 |
|
|
|
|
Functional assays: |
|
|
|
|
pSATA5 expression after stimulation |
|
|
|
Mycobacteria: Nontuberculous |
pSTAT3 expression after stimulation |
|
|
|
including BCG |
T cell proliferation assays by CFSE |
|
|
|
Protozoa:Pneumocystis jiroveci, |
Certain cytokine estimations: IL-10, |
|
|
|
Toxoplasma gondii, |
IL-12 and INF g |
|
|
|
Cryptosporidium parvum |
RBC ADA levels |
|
Antibody |
Upper and lower |
Bacteria:S. pneumoniae, |
B cells: |
|
deficiency |
respiratory tract, |
H. influenzae, M. catarrhalis,
|
1. B cell numbers: CD19, CD79a, CD20
|
|
|
GI tract, skin |
P. aeruginosa, S. aureus
|
2. Intracellular Btk expression |
|
|
infections, sepsis, |
N. meningitidis, M. pneumoniae |
3. Immunoglobulin estimation by |
|
|
meningitis |
|
nephelometry:Ig G, A, E,M |
|
|
|
|
Specific antibody responses |
|
|
|
Viruses: Enteroviruses |
|
|
|
|
Protozoa: Giardia lamblia |
|
|
Phagocytic |
Respiratory tract, Liver |
Bacteria: S.aureus, P. aeruginosa
|
Phagocytic Functions: |
|
defects |
or lung abscesses, GI |
Nocardia asteroids, S. typhi |
1. CD18, CD11 expression: |
|
|
diseases, urinary tract |
|
2. DHR |
|
|
problems |
Fungi: Candida, Aspergillus |
3.
NBT |
|
|
|
Mycobacteria: Nontuberculous
|
|
|
|
|
including BCG |
|
|
Complement |
Meningitis, systemic |
Bacteria: Streptococci, |
Functional hemolytic assay (CH50 and
|
|
deficiency |
bacterial infections |
H. influenzae, |
AH50 assays) and serum concentration
|
|
|
|
Neiserria |
measurement for complement components
|
|
|
|
Viruses: CMV, HSV |
|
Combined T and B Cell
Deficiency
There are 22 different groups
of diseases that have been included in this
category [1]. Severe combined immunodeficiencies
(SCID) like Adenosine deaminase (ADA)
deficiency, purine nucleotide phosphorylase
(PNP) deficiency, RAG1/2 deficiency,
a
chain deficiency, IL7Ra
deficiency, and JAK3 deficiency and combined
immunodeficiency (CID) like CD40 ligand
deficiency (X-linked hyper IgM) are some of the
common combined immunodefciencies. These
patients usually present within first six months
of life with failure to thrive, chronic diarrhea,
persistent oral thrush, skin rash, pneumonia,
and sepsis. Disseminated BCG infection is
commonly seen in patients with SCID. Similarly,
prolonged interstitial pneumonia of viral
etiology such as parainfluenza virus or
cytomegalovirus or Pneumocystis jerovici
is also common in patients with combined
immunodeficiency [6, 7].
Omenn syndrome is a rare
autosomal recessive disease usually presenting
in neonatal period, characterized by symptoms of
SCID associated with other findings like
erythroderma, lymphadenopathy,
hepatosplenomegaly and eosinophilia. Most of the
patients with PNP deficiency have neurological
problems including developmental delay,
hypertonia, spasticity, tremors, ataxia,
retarded motor development, behavioral
difficulties and varying degrees of mental
retardation. Characteristic abnormality in ADA
deficient SCID includes cupping at the end of
the ribs demonstrated on a chest radiograph.
They may also present with delayed development,
deafness and seizures.
Lymphopenia is commonly seen
with patients with SCID and requires further
evaluation for specific diagnosis. However
normal absolute lymphocyte count does not rule
out combined immunodeficiency, thus further
laboratory evaluation is required in case of
strong clinical suspicion. Neutropenia is seen
in many PIDs [8] including CD40L deficiency.
Predominant Antibody
Deficiency
This category includes 6
groups of diseases [1] of which X-linked
agammaglobulinemia (XLA) and common variable
immunodeficiency (CVID) are the commonest.
Patients with XLA typically present after 6-9
months of age when the level of protective
maternal IgG starts going down [9]. Recurrent
sinopulmonary infections due to S. pneumonia
or H. influenza, otitis media, and
septicemia are the most common clinical
manifestations. Less common manifestations
include enteroviral infections with resultant
chronic meningitis, dermatomyositis, and
rheumatoid like arthritis. Patients of CVID
usually present later in life that is after 5
years though some may present as early as 2
years of age. There is also an increased risk of
cancer in CVID cases predominantly with
lymphoreticular tumors and some patients can
also develop autoimmune diseases [10].
Other Well-defined PID
This category includes 9
different groups of diseases such as Ataxia
telengiectasia (AT), DiGeorge syndrome, Wiskott-Aldrich
syndrome (WAS) and hyper IgE syndrome (HIGE) [1].
Patients with AT or Nijmejen Breakage
syndrome can present at the age of 6 months to 5
years with gait abnormalities or
neurodevelopmental delay. Progressive cerebellar
ataxia with discrete or pronounced
telengiectasia involving the conjunctiva ears
and sometimes face are the classical findings in
ataxia-telengiectasia. Patients with DiGeorge
syndrome present in neonatal period. This defect
should be suspected in patients with cardiac
defects with hypoplastic thymus, hypocalcemia
and facial dysmorphism. Eczema in infancy and
recurrent staphylococcal skin boils and
pneumonia with pneumatocele formation are the
commonest presenting manifestations of HIES due
to STAT3 defect [11]. Patients with HIES due to
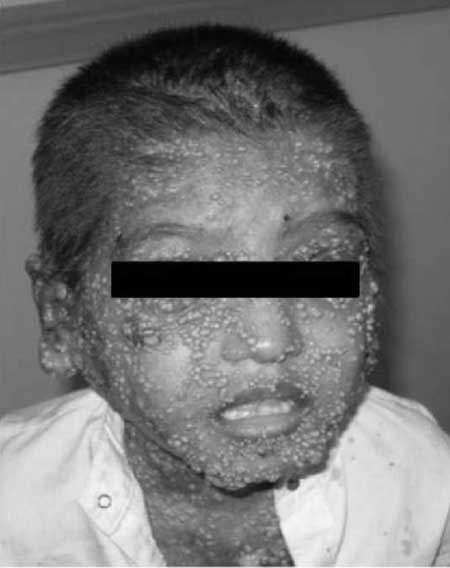
DOCK8 deficiency usually present with
disseminated moluscum contagiosum (Fig.1)
or disseminated viral warts [12]. Autosomal
dominant HIES is commonly associated with
multiple connective tissue and skeletal
abnormalities including scoliosis, hyper
extensibility, pathologic fractures, retained
primary dentition, craniosynostosis, and
vascular abnormalities [13]. Central nervous
system abnormalities are common in HIES.
Asymptomatic cerebral T2-weighted
hyper-intensities, increased prevalence of
lacunar infarcts, and increased Arnold Chiari 1
malformations are seen in the brain MRI of many
patients [14].
 |
|
Fig. 1 Patient
with HIES due to DOCK8 deficiency with
disseminated Molluscum contagiosum.
|
WAS patients present with
eczema, petechiae and recurrent sino-pulmonary
manifestations [15]. The incidence of EBV
associated lymphoma is also high in these
patients. Thrombocytopenia with low mean
platelet volume gives important clue for
diagnosis of WAS.
Phagocytic defects
This category of PID includes
5 groups of diseases [1] of which leukocyte
adhesion deficiency-I (LAD-I), chronic
granulomatous disease (CGD) and severe
congenital neutropenia (SCN) are some of the
common diseases. Patients with phagocytic
defects usually present in neonatal period.
Delayed separation of umbilical cord beyond 2
weeks along with omphalitis is suggestive of a
neutrophil disorder like LAD-I [16], SCN or CGD.
Patients with LAD-II have severe mental
retardation, short stature, a distinctive facial
appearance and the rare Bombay (Hh) phenotype.
Eczematous rash with deep seated abscesses is
associated with CGD. Infections due to S.
aureus, Burkholderia cepacia
and fungal infections (mainly
Aspergillus) are common in CGD [17].
Disseminated atypical
mycobacterial infection or BCGiosis or recurrent
salmonella infection in an otherwise well grown
individual leads to suspicion to Mendelian
susceptibility to mycobacterial diseases (MSMD)
due to type-I cytokine defects [18]. Persistent
neutrophilia even in the absence of active
infection is a common feature of LAD-I. In
severe congenital neutropenia child has
persistently low absolute neutrophil counts
(ANC) with elevated monocytes and eosinophils
counts. Cyclic neutropenia patients present with
drop in ANC every 3-4 weeks with fever,
infections and mouth ulcers.
Diseases of immune
dysregulation
Four groups of diseases are
included in this category [1]. Familial
Hemophagocytic Lymphohistiocytosis (FHL) that
includes Perforin deficiency, UNC13D (Munc13-4)
deficiency, Syntaxin 11 deficiency and STXBP2 (Munc
18-2) deficiency and Autoimmune
Lymphoproliferative syndrome (ALPS) are the most
common groups of diseases in this category.
Patients with diseases of perforin defect
usually present at less than six months of age.
Patients with Autoimmune Lymphoproliferative
syndrome (ALPS) present at the median age of
around 2 years with chronic nonmalignant
lymphadenopathy, splenomegaly and immune
cytopenias [19]. Mucocutaneous albinism is seen
with patients with Griselli syndrome and
Chediak-Highashi syndrome [20].
Patients with
hemophagocytic lymphohistiocytosis (HLH) (either
familial or associated with Chediak-Higashi or
Griscelli syndrome-II, X-linked
lymphoproliferative syndrome) are associated
with varying degrees of cytopenias with
hemophagocytosis seen either in the bone marrow
or rarely in the peripheral blood. Patients with
ALPS generally have elevated ALC with recurrent
non-malignant lymphadenopathy and
spleno-hepatomegaly. Patients with Evan’s
syndrome should be evaluated for underlying ALPS
as high proportion of these cases have FAS
gene mutation [21]. Autoimmune cytopenias
are also commonly seen in patients with immune
dysregulation, polyendocrinopathy, enteropathy
or X-linked (IPEX) syndrome. Hematolymphoid
malignancies are common with certain PID.
Patients with ALPS are at a higher risk of
developing the Non-Hodgkin and Hodgkin lymphoma.
EBV-associated lymphoma is common in patients
with XLP.
Complement deficiency
Patients with complement
deficiency present later in life usually after 5
years of age. Autoimmune disease and pyogenic
infections are often associated with a
deficiency of early components (complements 1-4)
of the classic pathway. Terminal complement
component deficiencies (complements 5-9) have
increased susceptibility to serious infections
from Neisseria species [10].
Complications such as recurrent pneumonia,
meningitis, and peritonitis are seen in
complement 3 deficiency.
Laboratory Approach to
Patients with PID
With wide array of assays
being available for evaluation of immune system,
it becomes difficult to choose an investigation
to be performed. The investigations are largely
guided by the clinical presentation of the
patient, the suspected immune defect and the
results of initial laboratory evaluation.
The most useful first-line
immunological investigations include a complete
blood count with a differential count on the
leucocytes and MPV, lymphocyte subset analysis,
serum immunoglobulin levels and Nitroblue
Tetrazolium test (NBT). The panel of antibodies
used for these purpose includes CD3, CD4, CD8,
CD56/16, CD19 and HLA-DR. It is aimed at
measuring the absolute and relative number of :
B cells (CD19+), T cells (CD3+), T-helper cells
(Th, CD3+/CD4+), T-cytotoxic cells (Tc,
CD3+/CD8+), Natural Killer (NK) cells
(CD3-/CD56+/CD16+), and Activated T cells
(CD3+/HLA-DR+).
It is very important to note
that the total lymphocyte numbers and T
lymphocyte subsets are age-dependent, being
markedly increased in newborns and young infants
and decreasing with age. In infants below 4
months of age, a CD4 count of <1000/mm3
is generally associated with impaired cellular
immunity, whereas the corresponding value is
<500/mm3
in children over 2 years of age and in adults
[22]. Immunosuppressive therapies like steroids
also significantly alter the values of T and B
cell subsets and should be interpreted
carefully.
The results of the initial
tests usually give an important clue to the
underlying immune defect. Patients with low T
cell counts are likely to have combined T and B
cell defects (CID). Patients with low or absent
B cell and low Immunoglobulin levels with normal
T cell fall in the category of predominantly
antibody deficiency. Patients with abnormal
neutrophil count or abnormal neutrophil function
suggest defects in the phagocytic system.
However, under these broad categories, there are
many subcategories or genetic defects and one
needs advanced laboratory tests available only
at specialized centers to come to a specific
diagnosis. Details of evaluation follow:
Suspected Combined T and
B-cell Immunodeficiency
Lymphocyte subset analysis is
abnormal in most cases of SCID and in many cases
of CID. SCID comprises of a group of inherited
disorders that characteristically show
abnormalities in T, B, and natural killer (NK)
cell function. These are categorized broadly as
T+ SCID and T-SCID depending on presence or
absence of the T cells.
There are many genetic
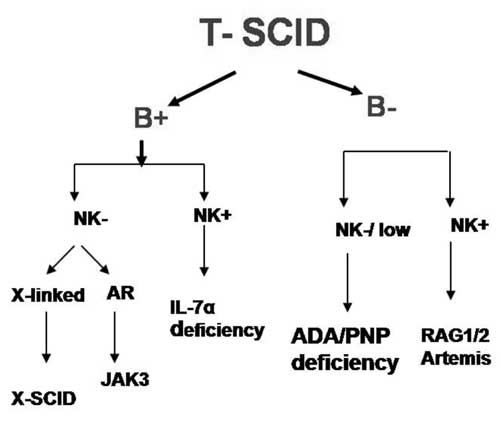
defects which can lead to T-SCID phenotype [23].
The B cells and NK cells count in these patients
give an important clue to the underlying
molecular defects (Fig. 2).
However, there is significant overlap between
these categories and hence specialized tests
like CD132 and CD127 expression, functional
studies like pSTAT5 activation in lymphocytes
after IL-2 stimulation, estimation of enzymes
like ADA and PNP in RBCs, radiation sensitivity
test, etc. are required for specific diagnosis.
 |
|
Fig. 2
Evaluation of patients with T- SCID.
|
Patients with normal T cell
numbers can still have CID. This is usually seen
with patients with Omenn syndrome, MHC-I or
MHC-II deficiency, ZAP 70 deficiency, etc. These
patients can be evaluated by doing T cell
proliferation assays (for evaluation of T cell
function), expression of HLA-DR on T and B cells
(for MHC class-II expression) and T cell
receptor (TCR) V-beta repertoire analysis (for
assessment of diversity of immune response).
Suspected B-cell Defect
Patients with suspected B
cell defects require estimation of B cell
numbers (CD19, CD20 and CD79a), and serum
immunoglobulin levels (IgG, IgA, IgM, IgE and
IgG subclasses). Patients with absent B cells
and markedly reduced Ig are suggestive of
agammaglobulinemia which can be X-linked (XLA)
or autosomal recessive agammaglobulinemia.
Patients with XLA will have absent or reduced
expression of protein Bruton Tyrosine Kinase
(BTK) with carrier mothers showing mosaic
pattern. Patients with reduced Ig with normal to
low B cells with abnormal specific antibody
responses suggest common variable
immunodeficiency (CVID). Patients with Hyper IgM
syndrome (HIGM) have markedly low IgG and IgA
with normal to elevated IgM levels. They can be
further evaluated by studying expression of CD40
and CD40L (CD154) expression on B cells and T
cells respectively. Patients with X-linked HIGM
will have abnormal CD 154 expression on T cells
after stimulation and carrier mothers will show
mosaic pattern.
Some patients with these
disorders may have normal or only modestly
reduced immunoglobulin levels; therefore, the
best approach for confirming a diagnosis of an
antibody-deficiency disorder is the measurement
of serum specific antibody titers (usually IgG)
in response to vaccine antigens. This approach
involves immunizing a patient with protein
antigens (e.g., tetanus toxoid) and
polysaccharide antigens (e.g., pneumococcus) and
assessing pre- and post-immunization antibody
levels. In many PIDs, antibody responses to
these antigens are diminished or even absent.
Suspected Phagocytic Defects
In a patient with suspected
phagocytic defect one must look at the absolute
neutrophil count (ANC). A patient with low ANC
with early neonatal presentation is suggestive
of severe congenital neutropenia (SCN).
Characteristically, there is marked monocytosis
with levels often two to four times that of
normal. There may be associated anemia and mild
thrombocytosis attributable to chronic
inflammation. Bone marrow examination shows the
presence of early precursor cells but very few
mature cells beyond the promyelocyte stage or ‘promyelocyte
arrest’. Patients with cyclic neutropenia have
oscillations of neutrophil count with a
periodicity of around 21 days. At the nadir,
neutrophil counts are generally less than 0.2×109
per L for 3-5 days, after which they rise
rapidly to levels near the lower limit of
normal, about 2×109
per L. Both SCN
and cyclic neutropenia commonly result from
mutations in neutrophil elastase gene (ELA-2).
Patients with suspected CGD
have normal or elevated ANC and can be diagnosed
by NBT and DHR test. These tests can also detect
carrier mothers in X-linked CGD. Final
confirmation of underlying defect can be done by
studying the intracellular expression of gp91
for X-CGD and p22, p67 or p47 for autosomal
recessive CGD followed by molecular analysis of
the affected gene. Patients with LAD-I can be
easily diagnosed by flowcytometric analysis of
CD18, CD11a, CD11b and CD11c expression on
peripheral blood leukocytes.
Suspected Immune
Dysregulation
There are two important
groups of disorders in this category.
Familial Hemophagocytic
Lymphohistiocytosis (HLH): HLH can result
from secondary causes like infections,
malignancy, rheumatic diseases or toxins or may
be due to inherited genetic defect leading to
impaired NK cell function. It is also important
to differentiate primary HLH from secondary HLH
as the patients with primary HLH are more likely
to relapse after therapy and require
hematopoietic stem cell transplantation for long
term survival. The diagnosis of HLH is often
difficult due to the rarity of the disease and
lack of a specific laboratory test, resulting in
under diagnosis. In an attempt to overcome these
difficulties, the FHL study group of the
Histiocyte Society has proposed diagnostic
guidelines for HLH (Table II)
[24]. The diagnosis of HLH is based on clinical
and laboratory criteria which involve complete
hemogram, bone marrow aspiration studies, liver
function tests, estimation of S bilirubin, S
ferritin, S triglyceride, S fibrinogen and sCD25
levels [25]. Further evaluation using NK cell
cytotoxicity assay, Perforin expression studies,
Granule release by NK cells and SAP and XIAP
expression on lymphocytes and MUNC 13-4 and
SYNTAXIN-11 by western blot help significantly
in diagnosis of genetic HLH.
TABLE II Diagnostic Criteria for HLH Proposed by Histiocyte Society
1. Molecular diagnosis of
hemophagocytic lymphohistiocytosis (HLH)
or X-linked lymphoproliferative syndrome
(XLP).
OR
2. at least 3 of 4:
a. Fever ³38.5°C
b. Splenomegaly
c. Cytopenias (minimum 2 cell lines
reduced)
Hemoglobin < 9g/dL (in infants < 4
weeks; Hemoglobin < 10g/dL;
Platelets < 100×103/mL;
Neutrophils < 1×103/mL
d. Hepatitis
3. And at least 1 of 4:
a. Hemophagocytosis
b. Ferritin > 500ng/mL
c. sIL2Ra (age-based)
d. Absent or very low NK function
4. Other results supportive of HLH
diagnosis:
a. Hypertriglyceridemia (>265 mg/dL)
b. Hypofibrinogenemia (<150 mg/dL)
c. Hyponatremia |
|
Source: ASH Education Book January
1, 2009 vol. 2009 no. 1 127-131. |
Autoimmune
lymphoproliferative syndromes (ALPS):
Autoimmune lymphoproliferative syndrome (ALPS)
is a disorder of lymphocyte homeostasis
characterized by non-malignant
lymphoproliferation autoimmunity mostly directed
toward blood cells and increased risk of
lymphoma. If ALPS is suspected based on clinical
findings, initial laboratory evaluation includes
flow cytometric analysis of peripheral blood
circulating TCR ab+
DNT cells and estimation of serum B12, soluble
FAS ligand (sFASL), interleukin (IL) -10 and
IL-18 levels. The recommended percentage of TCR
ab+DNT
cells required for a diagnosis is greater than
or equal to 1.5% of total lymphocytes or 2.5% of
T lymphocytes in the setting of normal or
elevated lymphocyte counts. The presence of
elevated TCR ab+
DNT cells coupled with high serum or plasma
levels of either IL -10, IL-18, (sFASL) or
vitamin B12 can accurately predict the presence
of germ line or somatic FAS mutations
[26].
Suspected Complement
Deficiency
In patients with suspected
complement deficiency, initial evaluation is
done with the CH50 (which tests the classical
and final lytic components except C9) and AH 50
(which tests alternative and final lytic
pathways) assays. These tests should be done in
laboratories with considerable experience of
these assays. To avoid misinterpretation due to
the possible effects of complement consumption
by immune complex formation, it is advisable
that the assays be performed when the patient
has completely recovered from immune complex
disease or infection. Both the tests require
blood to be taken atraumatically and serum be
separated within 1 hour and stored at -700C.
If either of these screening tests identifies
failure of a complement pathway on two
occasions, the specific component defect should
be determined.
Conclusion
Diagnosis of specific PID
from a large spectrum of disorders requires
expertise in clinical and laboratory evaluation.
Wide array of assays are available for
evaluation of immune system which help immensely
in the diagnosis of PIDs. Knowledge of clinical
presentation of these disorders, correct
interpretation of initial results of
immunophenotyping of lymphocytes is essential
for choosing the appropriate test for specific
diagnosis. There is very little data available
from India on PID. Being a country with the
second largest population in the world, we are
likely to have large number of patients with
PIDs. Recognizing a suspicious case of PID at a
regional hospital level is important to ensure
timely referral to a specialized centre for
diagnosis and treatment for these patients. We
need to create awareness about these disorders
and possibly establish more centers with
diagnostic facilities for their evaluation.
Contributors: MM has
conceived and written the manuscript. GK revised
the manuscript for important intellectual
content. MA compiled the data and helped in
manuscript writing. The final manuscript was
approved by all authors.
Funding: None;
Competing interests: None stated.
References
1. Al-Herz W, Bousfiha A,
Casanova JL, Chapel H, Conley
ME, Cunningham-Rundles C, et al. Primary
immunodeficiency diseases: an update on the
classification from the International Union of
Immunological Societies Expert Committee for
Primary Immunodeficiency. Frontiers in
Immunology. 2011;2:54.
2. Piguet D, Tosi C, Luthi
JM, Andresen I, Juge O. Redimune ((R)) NF
Liquid, a ready-to-use, high-concentration
intravenous immunoglobulin therapy preparation,
is safe and typically well tolerated in the
routine clinical management of a broad range of
conditions. Clin Exp Immunol.
2008;152:45-9.
3. Notarangelo LD, Forino C,
Mazzolari E. Stem cell transplantation in
primary immunodeficiencies. Curr Opin Allergy
Clin Immunol. 2006;6:443-8.
4. Subbarayan A, Colarusso G,
Hughes SM, Gennery AR, Slatter M, Cant AJ, et
al. Clinical features that identify children
with primary immunodeficiency diseases.
Pediatrics. 2011;127:810-6.
5. Slatter MA, Gennery AR.
Clinical immunology review series: an approach
to the patient with recurrent infections in
childhood. Clin Exp Immunol. 2008;152:389-96.
6. Winkelstein JA, Marino MC,
Ochs H, Fuleihan R, Scholl PR, Geha R, et al.
The X-linked hyper-IgM syndrome: clinical
and immunologic features of 79 patients.
Medicine (Baltimore). 2003;82:373-84.
7. Berrington JE, Flood TJ,
Abinun M, Galloway A, Cant AJ. Unsuspected
Pneumocystis carinii pneumonia at
presentation of severe primary immunodeficiency.
Arch Dis Child. 2000;82:144-7.
8. Bouma G, Ancliff PJ,
Thrasher AJ, Burns SO. Recent advances in the
understanding of genetic defects of neutrophil
number and function. Br J Haematol.
2010;151:312-26.
9. Wood P, Stanworth S,
Burton J, Jones A, Peckham DG, Green T, et
al. UK Primary Immunodeficiency Network.
Recognition, clinical diagnosis and management
of patients with primary antibody deficiencies:
a systematic review. Clin Exp Immunol.
2007;149:410-23.
10. Zi Yin E, Frush DP,
Donnelly LF, Buckleyl RH. Primary
immunodeficiency disorders in pediatric
patients: Clinical features and imaging
findings. Am J Roentgeno. 2001;176:1541-52.
11. Woellner C, Gertz EM,
Schäffer AA, Lagos M, Perro M, Glocker EO, et
al. Mutations in STAT3 and diagnostic
guidelines for hyper-IgE syndrome. J Allergy
Clin Immunol. 2010;125:424-32.
12. Zhang Q, Davis JC,
Lamborn IT, Freeman AF, Jing H, Favreau AJ,
et al. Combined immunodeficiency associated
with DOCK8 mutations. N Engl J Med.
2009;361:2046-55.
13. Buckley RH. The Hyper-IgE
syndrome. Clin Rev Allergy Immunol.
2001;20:139-54.
14. Freeman AF, Collura-Burke
CJ, Patronas NJ, Ilcus LS, Darnell D, Davis J,
et al. Brain abnormalities in patients
with hyperimmunoglobulin E syndrome. Pediatrics.
2007;119:e1121-5.
15. Albert MH, Notarangelo
LD, Ochs HD. Clinical spectrum, pathophysiology
and treatment of the Wiskott-Aldrich syndrome.
Curr Opin Hematol. 2010. Nov 11. [Epub ahead of
print]
16. Madkaikar M, Currimbhoy Z
, Gupta M, Desai M, Rao M. Clinical profile of
leukocyte adhesion deficiency. Indian Pediatr.
2011;49:1-4.
17. Holland SM. Chronic
granulomatous disease. Clin Rev Allergy Immunol.
2010;38:3-10.
18. Al-Muhsen S, Casanova JL.
The genetic heterogeneity of mendelian
susceptibility to mycobacterial diseases. J
Allergy Clin Immunol. 2008;122:1043-51.
19. Madkaikar M, Mhatre S,
Gupta M, Ghosh K. Advances in autoimmune
lymphoproliferative syndromes. Eur J Haematol.
2011;87:1-9.
20. Dessinioti C, Stratigos
AJ, Rigopoulos D, Katsambas AD. A review of
genetic disorders of hypopigmentation: lessons
learned from the biology of melanocytes. Exp
Dermatol. 2009;18:741-9.
21. Teachey D, Manno CS,
Axsom KM, Andrews T, Choi JK, Greenbaum BH,
et al. Unmasking Evans syndrome: T-cell
phenotype and apoptotic response reveal
autoimmune lymphoproliferative syndrome (ALPS).
Blood. 2005;105:2443.
22. Melaranci C, Ciaffi P,
Zerella A, Martinelli V, Cutrera R, Plebani A,
et al. T cell subpopulation in paediatric
healthy children: age-normal values. J Clin Lab
Immunol. 1992;38:143-9.
23. Walshe D, Gaspar HB,
Thrasher AJ, Cale CM, Gilmour KC. Signal
transducer and activator of transcription 5
tyrosine phosphorylation for the diagnosis and
monitoring of patients with severe combined
immunodeficiency. J Allergy Clin Immunol.
2009;123:505-8.
24. Henter J, Horne A, Aricó
M, Egeler RM, Filipovich AH, Imashuku S, et
al. HLH-2004: Diagnostic and therapeutic
guidelines for hemophagocytic
lymphohistio-cytosis. Pediatr Blood Cancer.
2006;48:124-31.
25. Janka GE. Familial and
acquired hemophagocytic lymphohistiocytosis. Eur
J Pediatr. 2007;166:95–100.
26. Caminha I, Fleisher TA,
Hornung RL, Dale JK, Niemela JE, Price S, et
al. Utilizing biomarkers to predict the
presence of FAS mutations in patients with
features of the autoimmune lymphoproliferative
syndrome. J Allergy Clin Immunol.
2010;125:946-9.
|
|
|
 |
|
