|
|
|
Indian Pediatr 2012;49: 488-490
|
 |
Novel STXBP2 Mutation Causing Familial
Hemophagocytic Lymphohistiocytosis
|
|
Rakhi Jain, Mammen puliyel, Prabhakar D Moses and
Elena Sieni*
From Department of Pediatrics, Christian Medical
College, Vellore, Tamilnadu, India and * Department of Pediatric
Hematology and Oncology, Anna Meyer Children’s Hospital, Florence,
Italy.
Correspondence to: Dr Prabhakar D Moses, Professor and
Head, Pediatrics Unit III, Christian medical College,
Vellore 632 004, Tamilnadu, India.
Email: child3@cmcvellore.ac.in
Received: June 6, 2011;
Initial review: June 27, 2011;
Accepted: September 27, 2011.
|
Familial Hemophagocytic Lymphohistiocytosis (FHL) is a rare autosomal
recessive disorder. Diagnosis is established in presence of genetic
mutation or positive family history in one of the siblings. Common
genetic mutations associated with FHL are mutations in gene PRF-1
(also known as FHL 2), UNC13D (FHL 3) and STX11
(FHL 4). Recently mutation in STXBP2 encoding syntaxin
binding protein 2 (Munc 18 -2) has been found to be associated with FHL
type 5. Here we describe the first reported Indian patient with
homozygous mutation in STX BP2 gene (c1697 G>A resulting
in amino acid change p.G566D) causing FHL 5.
Key words: Familial Hemophagocytic Lymphohistiocytosis, FHL 5,
STXBP2 mutation.
|
|
Familial hemophagocytic lymphohistiocytosis
(FHL) is a genetically heterogeneous immune disorder characterized
by widespread organ infiltration with activated macrophages and
lymphocytes. The clinical course usually starts in infancy and is
fatal unless treated with hematopoietic stem cell transplant.
According to the guidelines of the Histiocyte Society, diagnosis of
Hemophagocytic Lympho-histiocytosis (HLH) requires fulfillment of 5
out of 8 criteria – prolonged fever, splenomegaly, bicytopenia,
elevated triglycerides/low fibrinogen, increased ferritin,
hemophagocytosis in bone marrow, decreased NK cell cytotoxicity and
increased soluble CD25 [1]. Diagnosis of FHL is established in
presence of certain genetic mutations. Positive family history of
affected siblings strongly suggests the diagnosis of FHL. Disease
causing mutations in gene PRF-1encoding perforin, UNC13d
encoding MUNC 13-4, and STX11 encoding syntaxin11 have
been identified in approximately 80% of cases. Recently mutation in
STX BP2 encoding syntaxin binding protein 2 (Munc 18 – 2) has
been reported in a few patients. We report a patient with mutation
in STXBP2 gene from India.
Case Report
A 28 day-old girl presented with fever,
progressive abdominal distension and lethargy for seven days. She
was the first child born to third degree consanguineous parents.
During antenatal period mother gave history of fever and rash at
fifth month of gestation. At 36 weeks of gestation mother had
premature rupture of membranes and baby was delivered by caesarian
section for complicated breech. Baby cried at birth and birth weight
was 2.75 kg. Baby was admitted in NICU for suspected sepsis and
received 5 days of antibiotics. After discharge she was well for 14
days. At 21 days of life she developed low grade fever, progressive
abdominal distension and lethargy for which she was admitted
elsewhere and treated with intravenous cefotaxime and amikacin. In
view of decreasing hemoglobin and platelet count, she required
transfusion with blood products and antibiotics were changed to
vancomycin and meropenem. Bone marrow examination revealed
haemophagocytosis. She was given one dose of IV Immunoglobulin and
cyclosporine. Baby was brought to our hospital for further
management.
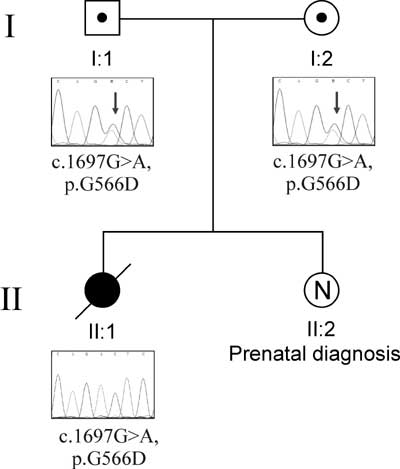 |
|
Fig. 1 Chromatophreograms of the
parents and the child highlighting the mutation in STXBP2
gene.
|
Physical examination revealed pallor, liver 6 cm
and spleen 4 cm palpable. Other system examination was unremarkable.
Investigations revealed haemoglobin of 8.7gm%, platelet count
41,000/cu mm, total leukocyte count 27,200/cu mm, serum ferritin
13280 ng/mL, triglycerides 458 mg/dL and undetectable fibrinogen.
Infection screen including blood culture, fungal culture, AFB
culture, TORCH infection and parvo virus serology were all negative.
A repeat bone marrow examination was done which showed increase in
reticuloendothelial activity with haemophagocytosis and no malignant
cells, thus fulfilling 6 out of 8 criteria for diagnosis of
Hemophagocytic Lymphohistiocytosis. Extracted DNA of the child and
the parents were sent for genetic analysis and she was started on
dexamethasone. The child improved, organomegaly decreased and blood
parameters normalized. Liver size at discharge was 2 cm, spleen 1cm.
However 12 days after discharge she was readmitted with fever and
poor feeding. Examination showed pallor and generalized skin
mottling, cold peripheries and feeble pulses. Liver was palpable 3cm
and spleen 2 cm. She was started on IV antibiotics, cyclosporine
6mg/kg/day was added after discussion with the hematologists. She
developed refractory shock and succumbed later.
Initial DNA analysis report revealed negative
mutation in PRF-1, UNC13D and STX11 genes but later
analysis revealed the new genetic mutation involving STXBP2
gene. The child was found to be homozygous for the following novel
mutation: c.1697G>A, resulting in amino acid change p.G566D. The
same mutation was found at heterozygous state in both the parents.
Mother is currently pregnant and prenatal DNA analysis of present
fetus showed normal karyotype and negative for mutation in the
STXBP2 gene.
Discussion
Four disease related genes have been identified
in FHL. In a cohort study using samples from West Asian countries,
mutations in already known genes (perforin, Munc13-4, and STX11)
were identified in 80% FHL patients, while STXBP2 mutation
accounted for 10% and the cause remained unknown for the remaining
10% of FHL cases (2). STXBP2 belongs to the Sec1/Mun18 family
of regulatory proteins involved in the assembly and disassembly of
SNARE (soluble N-ethylmaleimide sensitive factor attachment
protein [SNAP] receptor) complexes and intracellular trafficking.
STXBP2 is required for degranulation of NK cell cytotoxic
granules [3,4]. Mutation in STXBP2 results in defective
cytotoxic activity of NK cells.
Cetica, et al. [3] reported four patients
with STXBP2 mutations, originating from Italy, England, Kuwait and
Pakistan. Zur Stadt, et al. [5] reported 12 patients with
STXBP2 mutations from Turkey, Saudi Arabia, and Central Europe.
Meeths, et al. reported 11 patients from Pakistan, Denmark,
Netherlands, Norway and Russia and found that STX BP2
mutation is associated with a spectrum of clinical symptoms other
than those typically associated with HLH (colitis, bleeding
disorders, and hypogammaglobulinemia). This may be a reflection of
impaired expression and function of STXBP2 in cells other
than cytotoxic lymphocytes [6].
Our patient is the first reported Indian child
having novel mutation c.1697G>A in STXBP2 gene resulting in
amino acid change p.G566D and baby presented with typical
manifestations of HLH. Early genetic testing is needed to confirm
FHL as allogenic HCT is the only curative therapy. It further helps
in testing of at risk relatives, carrier testing, genetic counseling
and prenatal testing for pregnancies at risk if disease causing
mutation in family are known. As in our case we did prenatal
diagnosis for the second child which was negative for mutation in
STXBP2 gene.
Acknowledgement: Dr Maurizio Arico, Director
and Dr. Valentina Cetica, Department Pediatric Hematology Oncology,
Azienda Ospedaliero-Universitaria Meyer, Florence, Italy for
carrying out HLH mutation analysis.
Contributors: RJ: prepared the manuscript.
MP: involved in case management and sending mutation analysis. PDM:
edited the paper. ES: involved in genetic analysis. RJ and PDM:
revised the paper for important intellectual content. All authors
approved the final paper to be published.
Funding: None;
Competing interests: None stated.
References
1. Henter JI, Horne A, Arico M, Egeler RM ,
Filipovich AH , Imashuku S, et al. HLH-2004 : Diagnostic and
therapeutic guidelines for HLH. Pediatr Blood Cancer.
2007;48:124-131.
2. Cote M, Menager MM, Burgess A ,Mahlaoui N,
Picard C, Schaffner C, et al. Munc 18-2 deficiency causes FHL
5 and impaires cytotoxic granules exocytosis in patient NK cells. J
Clin Invest. 2009;119:3765-73.
3. Cetica V, Santoro A Glimou r KC, Seini E,
Beutel K, Arico M, et al. STX BP 2 mutation in children with
FHL 5. J Med Genet. 2010;47:595-600.
4. Sudhof TC, Rothman JE. Membrane fusion:
grappling with SNARE and SM proteins. Science. 2009;323:474-7.
5. ZurStadt U, Rohr J, Seifert W, Florian K,
Grieve S, Pagel J, et al. Familial Hemophagocytic
Lymphohistiocytosis type 5 is caused by mutations in Munc 18-2 and
impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482-92.
6. Meeths M, Entesarian M, Al-herz W, Nordenskjold
M , Bryceson Y, Henter JI, et al. Spectrum of clinical
presentation in familial hemophagocytic lymphohistio-cytosis type 5
patients with mutation in STXBP2. Blood. 2010;116:2635-43.
|
|
|
 |
|
