|
|
|
Indian Pediatr 2011;48:
457-466 |
 |
Holoprosencephaly: A Guide to Diagnosis and
Clinical Management |
|
Manu S Raam,† Benjamin D Solomon and Maximilian
Muenke
From Medical Genetics Branch, National Human Genome
Research Institute, National Institutes of Health, Bethesda, MD,
United States; and †HHMI-NIH Research Scholars Program, Howard Hughes
Medical Institute,
Chevy Chase, MD, United States.
Correspondence to: Maximilian Muenke, Building 35, Room
1B-203, 35 Convent Drive MSC 3717,
Bethesda, MD 20892-3717, United States.
Email: mamuenke@mail.nih.gov
|
Context: Holoprosencephaly affects 1 in 8,000 live births and is the
most common structural anomaly of the developing forebrain, resulting in
facial dysmorphism, neurologic impairment, and additional clinical
sequelae. Given the increasing relative contribution of genetic diseases
to perinatal morbidity and mortality in India, proper recognition and
management of holoprosencephaly can improve care for a significant number
of affected Indian children.
Evidence Acquisition: We used the PubMed database
(search terms: "holoprosencephaly," "HPE," "holoprosencephaly India") and
cross-referenced articles regarding holoprosencephaly, using our research
group’s extensive experience as a guide for identifying seminal papers in
the field.
Results: Holoprosencephaly is classified into four
types based on the nature of the brain malformations as seen on
neuroimaging and/or pathologic examination, with typically recognizable
craniofacial phenotypes. Despite the identification of several genetic
loci and other etiologic agents involved in pathogenesis, additional
causes are elusive. Moreover, satisfactory explanations for phenomena such
as incomplete penetrance and variable expressivity are lacking.
Conclusions: For each patient, pediatricians should
follow a diagnostic protocol including dysmorphology examination, complete
family history and ascertainment of risk factors, and neuroimaging. Many
medical issues, including hypothalamic dysfunction, endocrinologic
dysfunction, motor impairment, respiratory issues, seizures, and
hydrocephalus should be prioritized in management. Pediatricians should
work with genetic specialists to identify syndromic forms and to perform
cytogenetic investigation, molecular screening, and genetic counseling in
order to fully characterize prognosis and recurrence risk.
Key words: Diagnosis, Genetics, Holoprosencephaly, Management,
Review.
|
|
H
oloprosencephaly is the most
common structural anomaly of the developing forebrain, resulting from incomplete
midline cleavage of the prosencephalon and associated with neurologic
impairment and dysmorphism of the brain and face. Studies in humans and
animals suggest that the defects associated with holoprosencephaly occur
at the human equivalent of approximately two to three weeks
post-conception [1], indicating that holoprosencephaly is a disorder of
gastrulation. Holoprosencephaly occurs rather frequently, having been
observed in 1:250 conceptuses [2]; due to a high rate of fetal demise, the
birth prevalence is 1:8000 live births [3]. As subsequently discussed in
greater detail, India’s large population size, unique population
structure, and perinatal morbidity and mortality patterns indicate that
proper recognition and management of congenital disorders like
holoprosencephaly by pediatricians and medical geneticists can improve
healthcare for a sizeable number of Indian children.
Our research group, located at the National Human
Genome Research Institute (National Institutes of Health) in the United
States, has extensive clinical and research experience with
holoprosencephaly, and routinely works with patients and families affected
by holoprosencephaly, as well as with blood samples sent to us from within
and outside the US. In the following text, we aim to provide the
practicing Indian pediatrician with information regarding cardinal
clinical and genetic concepts regarding holoprosencephaly, with a special
emphasis on clinical management and molecular diagnostic options available
to enhance care of Indian children with the condition.
Epidemiology and Implications
Significant variation from the base prevalence of
1:8000 live births has not been observed among different international
populations in several multi-center studies. In the United States,
seemingly higher prevalences have been reported in Hispanic,
African-American, and Pakistani ethnicities, likely attributable to
decreased prenatal diagnosis and termination rates in these groups [4].
This situation may be extrapolated to other countries, including India; as
in any population, variable levels of knowledge regarding
holoprosencephaly and reduced access to prenatal healthcare in specific
communities may lead to higher apparent prevalences and suboptimal
clinical management.
There is a paucity of specific information regarding
Indian patients with holoprosencephaly in the literature; the largest case
series of Indian patients with holoprosencephaly consisted of 13 patients
and was described in 2004 [5]. Nevertheless, the lack of such descriptions
is not likely to be due to a reduced number of Indian patients with the
condition. In fact, large family sizes and high rates of consanguineous
marriages in India lead one to expect increased occurrence of certain
genetic disorders [6], and the enormous Indian population size translates
to a large number of infants (495,000 per year) who are affected by all
genetic disorders [7]. Given the increasing relative contribution of
genetic disease to perinatal morbidity and mortality [7], it is reasonable
to expect that an Indian pediatrician in a large city would encounter and
be required to significantly manage critically ill patients with
holoprosencephaly.
Classification Schema
Holoprosencephaly is classically divided into four
types, based on the degree of nonseparation of the prosencephalon [8,9].
These types, in order of increasing cortical separation, include the
alobar form, characterized by diffuse cortical nonseparation; the
semilobar form, characterized by non-separation of the frontal lobes; the
lobar form, characterized by nonseparation of the basal aspect of the
frontal lobes; and the middle interhemispheric variant, characterized by
nonseparation of the posterior frontal and parietal lobes [10] (Fig.1).
Additional nuances specific to each type are described in Table
I. As further described in the section on clinical management,
severity of craniofacial malformations and prognosis tend to correlate
with the degree of nonseparation: the alobar form is the most severe in
terms of both craniofacial malformations and neurologic impairment; the
semilobar form is characterized by milder or absent craniofacial
malformations, but persistence of severe motor abnormalities; and, the
lobar and middle interhemispheric variant forms are comparatively mild,
both in terms of craniofacial malformations and neurologic impairment
[10]. Finally, very mildly affected "microforms" have been described,
wherein individuals may display subtle craniofacial features including
microcephaly, hypotelorism (closely spaced eyes), and single maxillary
central incisor but typically do not demonstrate obvious radiologic
evidence of nonseparation or severe neurologic impairment [11].
Table I
Descriptions Of Typical Brain Findings In Each Of The Types Of Holoprosencephaly
|
|
Alobar |
Semilobar |
Lobar |
MIHV* |
Microform |
|
Interhemispheric separation |
Complete or near-complete nonseparation,
with absent falx cerebri |
No anterior separation, some posterior
separation |
Nonseparation of only the most rostral/ventral
frontal neocortex, with hypoplastic falx cerebri |
Nonseparation of posterior frontal and
parietal lobes |
No interhemispheric fusion |
|
Corpus callosal characteristics |
Absent corpus callosum |
Absent anterior corpus callosum |
Absent corpus callosum in affected region |
Absent body of the corpus callosum |
May have subtle defects |
|
Additional findings |
Absent olfactory bulbs, fused deep gray
nuclei, and single midline monoventricle |
Absent or hypoplastic olfactory bulbs,
fused deep gray nuclei, and absent anterior horns of lateral
ventricles and septum pellucidum |
Hypoplastic olfactory bulbs, hypoplastic
falx cerebri, and azygous anterior cerebral artery |
Frequent fusion of thalami and caudate
nuclei, Gray matter heterotopias, cortical dysplasia, and
Azygous anterior cerebral artery |
May have subtle midline brain defects |
|
* MIHV: middle interhemispheric variant; Reproduced with permission
from reference 10. |
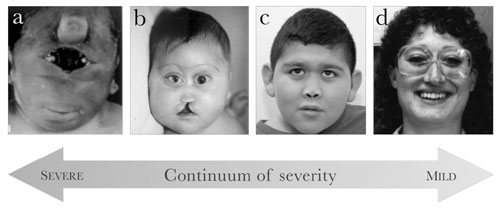 |
|
Fig. 1 Craniofacial phenotypes in patients
with holo-prosencephaly. From left to right: (a)
synophthalmia and a proboscis in a patient with alobar
holoprosencephaly; (b) severe hypotelorism, flat nasal
bridge, bilateral colobomas, and midline cleft lip and palate in a
patient with alobar holoprosencephaly; (c)
hypotelorism, flat nasal bridge, and closely spaced nostrils in a
patient with lobar holoprosencephaly; (d) hypotelorism, sharp nasal
bridge, and single maxillary central incisor in an individual with a
microform of holoprosencephaly. (Adapted from [20] and [25] with
permission from Nature Publishing Group and BMJ Publishing Group,
Ltd., respectively.) |
Craniofacial Findings
In most but not all cases, craniofacial manifestations
tend to follow DeMyer’s 1964 maxim, "the face predicts the brain" [12]. In
other words, the severity of the craniofacial phenotype tends to mirror
the severity of the brain malformations and correlates inversely with
survival [13] (Fig.2). The most severe facial phenotypes
include pronounced micro-cephaly, cyclopia (single, centrally placed eye),
synophthalmia (partial union of the two eyes in the center of the face),
and a proboscis (a tube-like nasal appendage with a single nostril located
above the ocular region) [13]. Less severe facial phenotypes can include
microcephaly (except in cases of hydrocephalus, which can cause
macrocephaly), hypotelorism, midface hypoplasia with a flat nasal bridge,
cleft lip and/or palate, ocular colobomas, and a single maxillary central
incisor [13]. Individuals with microforms of holoprosencephaly, usually
identified as relatives of probands with frank holoprosencephaly, have
isolated craniofacial findings without the classic clinical issues and
neurologic impairment seen in holoprosencephaly [11,13]. Conversely,
individuals with mutations in ZIC2, one of the genes implicated in
select cases of holoprosencephaly, present an exception to the "face
predicts the brain" maxim, as these patients have severe holoprosencephaly,
neurologic impairment, and characteristic clinical sequelae, but have a
much milder facial phenotype than that of other patients [13,14].
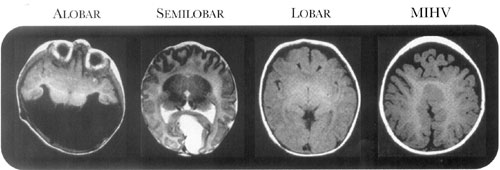 |
|
Fig 2. Axial sections through cranial MR
images of patients with holoprosencephaly, distinguished by type.
MIHV: middle interhemispheric variant. (Adapted from [32] with
permission from Elsevier.) |
Etiology and Molecular Genetics
The etiology of holoprosencephaly is extremely
heterogeneous and is still being elucidated. With varying levels of
evidence, a number of environ-mental factors and teratogens have been
suggested, including maternal diabetes (infants born to diabetic mothers
have a 200-fold risk of holoprosencephaly), ethanol, cytomegalovirus
infection, salicylates, anti-epileptic medications, retinoic acid, and
maternal hypocholesterolemia [15,16]. Genetic causes have also been
implicated, based on familial occurrences of holoprosencephaly, the
presence of known syndromes and associations including holoprosencephaly,
and the nonrandom nature of chromosomal aberrations in patients with
holoprosencephaly [16]. Between 18%-25% of live births affected by
holoprosencephaly have a recognizable monogenic syndrome, including Smith-Lemli-Opitz
syndrome (MIM #270400), Pallister-Hall syndrome (MIM #146510), and
Rubinstein-Taybi syndrome (MIM #180849) [16]. Chromosomal anomalies have
been implicated in 24-45% of live births affected by holoprosencephaly
[16-18], most frequently numeric anomalies in chromosomes 13, 18, and 21
[19] and structural anomalies involving 13q, 18p, 7q36, 3p24-pter, 2p21,
and 21q22.3 [16]. Intragenic mutations in four genes have also been firmly
established as increasing susceptibility to holoprosencephaly: SHH
(7q36) (20-22), SIX3 (2p21) (23-25), ZIC2 (13q32) (14, 26),
and TGIF (18p11.3) [27]. While testing for mutations in these four
genes has led to significant diagnostic advancements and implications for
patient care, 75% of chromosomally normal patients with holoprosencephaly
do not have identified mutations in any screened genes [28], indicating
the need to identify additional susceptibility genes.
The genetics of holoprosencephaly are such that
multiple affected individuals can present with holoprosencephaly within
the same family, but incomplete penetrance and variable expressivity lead
to tremendous intrafamilial phenotypic variability [29]. A related
observation is that individuals with certain chromosomal aberrations and
intragenic mutations associated with holoprosencephaly may not actually
have holoprosencephaly in all cases: only 50% of patients with deletions
in 7q36, including SHH, have holoprosencephaly, and only 10% with
deletions in 18p, including TGIF, do so [30]. Thus,
holoprosencephaly, like many other entities considered to be "simple"
Mendelian dis-orders, is characterized by complex traits that are not
reliably predicted by the presence of a single mutation [31].
Diagnosis
A recommended protocol for clinical and molecular
diagnosis in patients with holoprosencephaly is provided in Fig.
3. The diagnostic process is typically initiated by abnormal prenatal
brain imaging, positive physical examination findings, and/or positive
family history. Whenever possible, a thorough dysmorphology examination
and an inter-view to determine risk factors and family history should be
obtained. Ascertainment of the specific neurologic findings and
holoprosencephaly type in each patient, via brain imaging, is essential to
proper counseling of the patient and his/her family, given their effect on
prognosis. MR (magnetic resonance) imaging provides the highest quality
data for this purpose, allowing detailed analysis of cortical white matter
and structural abnormalities of the deep gray nuclei [10], although
logistic issues and the risks of the sedation required in neurologically
impaired patients can make this impractical. If MR imaging cannot be
performed, other options include ultra-sound, which can be performed while
the fontanelles are patent, and CT (computed tomography) imaging, which
carries risks associated with radiation expo-sure. If a patient is found
to have microcephaly, a large dorsal cyst, or rapidly enlarging head size,
serial imaging is indicated [32].
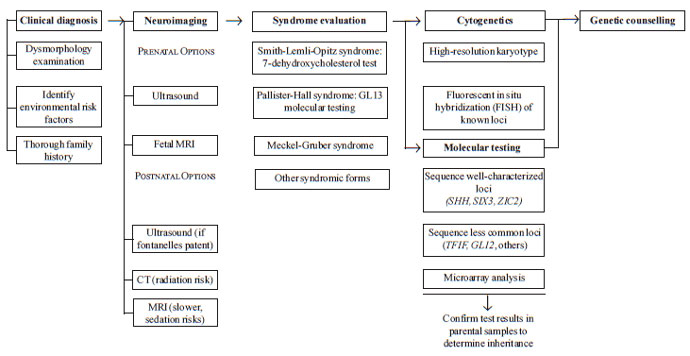 |
|
Fig 3 Recommended clinical protocol
for diagnosing and elucidating causes of holoprosencephaly in
patients. Each of the six major steps is medically indicated; within
each step, bolded items are medically indicated or preferred, while
others are performed if suggested by the clinical characteristics of
the patient or at the discretion of the clinical laboratory. See
text for more details. |
Prenatally, providing an early date of diagnosis is
important from both scientific and psychologic points of view, because the
severity of malformations leads to emotional effects among family members
and may include consideration of pregnancy termination [33,34]. Prenatal
ultrasound of the face and falx cerebri can be used to diagnose alobar and
semilobar holoprosencephaly as early as the first trimester [10,33], while
fetal MRI provides more sensitive diagnosis for milder forms of
holoprosencephaly during the third trimester [35]. Ultra-sound remains
the gold standard due to its relative imperviousness to maternal obesity,
fetal position, bone reverberation, and oligohydramnios [34]. In a recent
study comparing ultrasound-based diagnosis to postmortem autopsy findings,
autopsy confirmed the prenatal diagnosis of holoprosencephaly in 17/21
cases, with two patients unable to receive a precise pathological
diagnosis due to extensive severity of malformations, and two additional
patients found to have severe complex brain and facial malformations other
than holoprosencephaly [34]. Ultrasound is not completely accurate in
determining holoprosencephaly type: in 7/17 cases, the holoprosencephaly
type determined through prenatal diagnosis differed from that determined
via postmortem autopsy [34]. In families with an existing child with
holoprosencephaly and an identified disease-causing mutation, prenatal
molecular diagnosis is possible, although presence of the mutation does
not necessarily portend holoprosencephaly [35].
Prognosis
Survival rates vary in each type of holoprosencephaly,
but in general, mortality correlates positively with the severity of the
brain malformation and, by extension, severity of the facial phenotype
[13]. Of children with alobar holoprosencephaly, those with severe facial
anomalies such as cyclopia and proboscis rarely survive the immediate
postnatal period, while those with less severe facial malformations can
survive for months or, in a minority of cases, longer than one year [36].
In very rare instances, survival into the twenties has been observed
(authors’ own experience). In contrast to most children with alobar
holoprosencephaly, children with holoprosencephaly types other than alobar
may more often survive into adulthood [36]. Frequent causes of death
include respiratory infections, dehydration secondary to uncontrolled
diabetes insipidus, intractable seizures, and sequelae of brainstem
malfunction, including aberrant control of respiration and heart rate
[36].
As with survival, developmental outcomes generally
correlate with the severity of the brain malformation, although again,
tremendous variability can occur. Children with alobar holoprosencephaly
may develop to a stage equivalent to that of a healthy, early infant:
while they may track objects or sounds, they typically cannot speak words,
sit without assistance, or reach for objects [37]. In contrast, some
children with semilobar holoprosencephaly can develop receptive language
skills, and while speech is still frequently impaired, they can
communicate through eye movements, gestures, or other non-verbal
communication systems, and may be socially engaging [37]. The severe motor
impairment observed in alobar and semilobar holoprosencephaly is less
frequently seen in the lobar type and the middle interhemispheric variant;
patients with the latter forms may walk with assistance, adequately
control their limbs, and even speak words or sentences [37]. The enhanced
vocal communication in these patients may be explained by more complete
separation of the deep gray nuclei, but because separation of the deep
gray nuclei does not appear to correlate with social awareness, visual
attention, and auditory comprehension, differences in those constructs may
be caused by structural changes in different regions [38]. The Carter
Neurocognitive Assessment (CNA) may be useful to clinicians for assessing
cognitive function in children with more severe impairment [38].
Clinical Management
Due to the medial and rostral location of the
hypothalamus, nonseparation of the hypothalamus occurs frequently, leading
to a variety of issues involving homeostatic and hypothalamic-pituitary
endocrine functions [39]. One disturbed homeostatic function is body
temperature regulation, which is significant for two reasons: first,
ascertainment of baseline body temperature helps identify abnormal
deviations in temperature due to infections or other causes of morbidity;
second, temperature instability in itself can cause morbidity and organ
dysfunction if the core temperature falls below 34ºC or rises above 40ºC
[37]. Other impaired homeostatic functions include thirst, appetite, and
sleep-wake cycles, disturbances of all of which can pose significant
problems for caregivers [37].
From an endocrinologic perspective, dysfunction of the
posterior pituitary, in the form of central diabetes insipidus, is much
more commonly observed than anterior pituitary insufficiency [39,40],
typically manifesting with polyuria, dehydration, hypernatremia, and
decreased urine osmolarity [40]. The severity of diabetes insipidus
generally correlates with the degree of hypothalamic nonseparation but
not with pituitary gland defects observed via imaging [40]. Due to the
high incidence of posterior pituitary dysfunction, and because diabetes insipidus in these patients may be asymptomatic, routine screening of
electrolyte levels for evidence of posterior pituitary endocrinopathies is
recommended in all patients, with repeated testing even in the event of
an initial negative result and also in the acute setting [37,40]. Anterior
pituitary issues, occurring with lower frequency than posterior pituitary
issues, include hypothyroidism, hypocortisolism, growth hormone
deficiency, and multiple pituitary hormone deficiency [40]. As signs of
hypothyroidism and hypocortisolism can be difficult to distinguish from
those seen classically in holoprosencephaly, and because the effects of
those endocrinologic deficiencies can be life-threatening, we recommend
basic screening evaluations in all patients, but with in-depth stimulation
tests only if clinical suspicion is high.
Motor impairment in holoprosencephaly generally
manifests as hypotonia, dystonia, and/or spasticity, frequently requiring
pharmaceutical inter-ventions such as intrathecal baclofen pumps and oral
trihexyphenidyl, as well as physical and occupational therapy and
surgical interventions [37]. One of the most detrimental effects of motor
impairment is oromotor dysfunction, which significantly com-pounds the
thirst and appetite disturbances resulting from hypothalamic dysfunction,
and may also exacerbate unique feeding challenges secondary to cleft lip
and palate [37]. Children with such issues frequently develop
oropharyngeal dysphagia and respiratory symptoms related to aspiration and
difficulty managing secretions, compromising oral intake and increasing
the risk of respiratory infections. Additional respiratory issues can
include chronic lung disease with decreased pulmonary reserve and chronic
inflammation. A gastrostomy tube is placed in many children with oromotor
dysfunction to address these issues. Gastrointestinal issues related to
poor nervous regulation, including poor gastric and colonic motility and
gastro-esophageal reflux, can still impair feeding despite placement of a
gastrostomy tube, sometimes indicating medications and anti-reflux
procedures [37].
Finally, the nature of the brain malformation may
predispose patients to seizures and/or hydrocephalus. Seizures occur in
approximately half of the patients [39], most commonly complex partial
seizures, and typically develop during infancy [37]. In addition, "epileptiform"
activity has been noted on electroencephalograms (EEGs) of some patients
without overt clinical seizures [41], suggesting that routine EEG
screening of patients may be useful. Of patients with recurring seizures,
most are managed with one or two antiepileptic medications; intractable
seizures occur in one-third to one-half, typically in patients with more
severe cortical malformations [37,39]. As seizure triggers can include
fluid and electrolyte imbalances from diabetes insipidus, proper
management of seizures requires consideration of endocrinologic issues
[37]. Hydrocephalus is another common finding that depends on the specific
brain malformation, correlating highly with thalamic nonseparation and the
presence of a dorsal cyst; it is thought to result from blocked
cerebrospinal fluid egress from the third ventricle [42]. Because
holoprosencephaly typically results in microcephaly, hydrocephalus should
be suspected in patients with normal head sizes or macrocephaly and
followed using serial head circumference measurements and ultrasound
imaging [37]. Placing a cerebrospinal fluid shunt, while taking particular
care to avoid overdrainage, can improve developmental outcomes, improve
other issues, and reduce macrocephaly [37].
Thus, diverse clinical sequelae can result from a
primary insult of holoprosencephaly. Clinicians should have a low
threshold for testing for these sequelae, as specific abnormalities are
difficult to predict in advance and may be challenging to diagnose.
Further Steps to Elaborate Genetic Causes and
Inheritance
Indian pediatricians are well-equipped to clinically
diagnose holoprosencephaly and to manage the clinical sequelae of the
condition, but full benefit to the patient and his/her family cannot be
achieved without genetic investigation. We recognize that there are many
barriers to the consistent application of genetic testing and
interpretation to each Indian patient with holoprosencephaly, as the
current state of medical genetics in India leaves many clinicians without
formal training in genetics and easy access to affordable genetic testing
laboratories [43]. Nevertheless, for a proper discussion with the family
regarding etiology and recurrence risk, pediatricians should seek out
genetic specialists within or outside India who are familiar with
holoprosencephaly and discuss the feasibility of genetic testing with
them. Here, we briefly discuss what is needed so that the pediatrician may
be familiar with the process.
As previously mentioned, holoprosencephaly frequently
occurs as part of a syndrome, and additional diagnostic steps should be
undertaken if the patient is clinically suspected to be affected by one of
these syndromes. For instance, patients suspected to have Smith-Lemli-Opitz
syndrome should have total cholesterol and 7-dehydro-xycholesterol levels
checked for a decrease and an increase outside the normal range,
respectively [44].
To determine genetic causes of holoprosencephaly in
each patient, a combination of cytogenetic and molecular testing is
recommended. Due to the high incidence of chromosomal anomalies, a
high-resolution karyotype at the 550 band level or greater is indicated in
all patients. Direct DNA sequencing of SHH, ZIC2, and
SIX3 is also indicated, due to the high prevalence of intragenic
mutations in those genes [45]. DNA sequencing results should be compared
to analyses of biologic effects and results of functional studies [22,24]
for each potential mutation, which are essential to help determine the
true pathogenic import of each variant. Routine sequencing of minor loci
is not performed unless indicated by specific observations in a patient:
for instance, pituitary abnormalities in a patient with holoprosencephaly
may warrant molecular testing of GLI2 [45] due to an emerging
genotype/phenotype correlation [13]. Microarray analysis, including
array-based comparative genomic hybridization (array CGH, or aCGH) and
single nucleotide polymorphism (SNP) arrays, is a relatively new molecular
technique that allows for identification of deletions and duplications at
resolutions far exceeding that of a karyotype, but currently, the novelty
of this technique indicates that logistical and financial barriers, as
well as the inadequacy of information allowing us to separate benign copy
number variants from pathogenic deletions and duplications [46], may need
to be addressed before the technique is used more routinely.
All of the information gathered through the steps
outlined above is necessary for proper genetic counseling, the need for
which is established by the poor prognosis in the most severely affected
patients and the relative uncertainty of each patient’s severity a
priori due to the extreme phenotypic variability of the condition.
Effective genetic counseling takes into account the inconsistency of
strict genotype-phenotype correlations for each identified genetic
variant, indicating the need for caution while interpreting molecular
results. Although medical genetics may not be a particular physician’s
area of expertise, we urge pediatricians to familiarize themselves with
the above recommendations and to correspond with medical geneticists, so
that the quality of genetic counseling can be enhanced and further
morbidity and mortality related to holoprosencephaly can be ameliorated.
Contributors: All authors participated in
the critical editing of this review article.
Competing interests: No authors have any
financial/personal disclosures or competing interests.
Funding: Support was provided in part
by Division of Intramural Research at the National Human Genome
Research Institute (National Institutes of Health, Department of Health
and Human Services, United States of America).
|
Key Messages
• Holoprosencephaly is characterized by failure
of the prosencephalon to divide into complete hemispheres, and is
associated with facial dysmorphism and neurologic impairment.
• Essential components of diagnosis include a
thorough interview to determine family history and teratogenic
exposures, dysmorphology exam, and neuroimaging, which is critical
for prognosis determination.
• Medical management should focus on
hypothalamic and endocrinologic dysfunction, motor and
developmental impairment, respiratory issues, seizures, and
hydrocephalus.
• Pediatricians should follow up medical management by
collaborating with a genetic specialist, with the aim of
performing genetic testing, determination of associated syndromes,
and genetic counseling.
|
References
1. O’Rahilly R, Müller F. Interpretation of some median
anomalies as illustrated by cyclopia and symmelia. Teratology.
1989;40:409-21.
2. Matsunaga E, Shiota K. Holoprosencephaly in human
embryos: epidemiologic studies of 150 cases. Teratology. 1977;16:261-72.
3. Leoncini E, Baranello G, Orioli IM, Annerén G,
Bakker M, Bianchi F, et al. Frequency of holoprosencephaly in the
International Clearinghouse Birth Defects Surveillance systems: searching
for population variations. Birth Defects Res A. 2008;82:585-91.
4. Orioli IM, Castilla EE. Epidemiology of
holoprosen-cephaly: prevalence and risk factors. Am J Med Genet C Semin
Med Genet. 2010;154C:13-21.
5. Thakur S, Singh R, Pradhan M, Phadke SR. Spectrum of
holoprosencephaly. Indian J Pediatr 2004;71:593-7.
6. Verma IC. Medical genetics in India. Indian J
Pediatr. 1986;53:437-40.
7. Verma IC. Burden of genetic disorders in India.
Indian J Pediatr. 2000;67:893-8.
8. DeMyer W, Zeman W. Alobar holoprosencephaly (arhinencephaly)
with median cleft lip and palate: clinical, electroencephalographic and
nosologic considerations. Confin Neurol. 1963;23:1-36.
9. Barkovich AJ, Quint DJ. Middle interhemispheric
fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol.
1993;14:431-40.
10. Hahn JS, Barnes PD. Neuroimaging advances in
holoprosencephaly: refining the spectrum of the midline malformation. Am J
Med Genet C Semin Med Genet. 2010;154C:120-32.
11. Solomon BD, Lacbawan F, Jain M, Domené S, Roessler
E, Moore C, et al. A novel SIX3 mutation segregates with
holoprosencephaly in a large family. Am J Med Genet A. 2009;149A:919-25.
12. DeMyer W, Zeman W, Palmer CG. The face predicts the
brain: diagnostic significance of median facial anomalies for
holoprosencephaly (arhinencephaly). Pediatrics. 1964; 34:256-63.
13. Solomon BD, Mercier S, Vélez JI, Pineda-Alvarez DE,
Wyllie A, Zhou N, et al. Analysis of genotype-phenotype
correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet.
2010;154C:133-41.
14. Solomon BD, Lacbawan F, Mercier S, Clegg NJ,
Delgado MR, Rosenbaum K, et al. Mutations in ZIC2 in human
holoprosencephaly: description of a novel ZIC2-specific phenotype
and comprehensive analysis of 157 individuals. J Med Genet.
2010;47:513-24.
15. Johnson CY, Rasmussen SA. Non-genetic risk factors
for holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:73-85.
16. Muenke M, Beachy PA. Holoprosencephaly. In:
Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and
Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001.
p.6203-30.
17. Croen LA, Shaw GM, Lammer EJ. Holoprosencephaly:
epidemiologic and clinical characteristics of a California population. Am
J Med Genet. 1996; 64:465-72.
18. Olsen CL, Hughes JP, Youngblood LG, Sharpe-Stimac
M. Epidemiology of holoprosencephaly and phenotypic characteristics of
affected children: New York State, 1984-1989. Am J Med Genet.
1997;73:217-26.
19. Solomon BD, Rosenbaum KN, Meck JM, Muenke M.
Holoprosencephaly due to numeric chromosome abnormalities. Am J Med Genet
C Semin Med Genet. 2010; 154C:146-48.
20. Roessler E, Belloni E, Gaudenz K, Jay P, Berta P,
Scherer SW, et al. Mutations in the human Sonic Hedgehog
gene cause holoprosencephaly. Nat Genet. 1996;14:357-60.
21. Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW,
de Die-Smulders C, et al. The mutational spectrum of the Sonic
Hedgehog gene in holoprosencephaly: SHH mutations cause a
significant proportion of autosomal dominant holoprosencephaly. Hum Mol
Genet. 1999;8: 2479-88.
22. Roessler E, El-Jaick KB, Dubourg C, Vélez JI,
Solomon BD, Pineda-Álvarez DE, et al. The mutational spectrum of
holoprosencephaly-associated changes within the SHH gene in humans
predicts loss-of-function through either key structural alterations of the
ligand or its altered synthesis. Hum Mutat. 2009;30:E921-35.
23. Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire
T, Richieri-Costa A, et al. Mutations in the homeodomain of the
human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196-8.
24. Domené S, Roessler E, El-Jaick KB, Snir M, Brown JL,
Vélez JI, et al. Mutations in the human SIX3 gene in
holoprosencephaly are loss of function. Hum Mol Genet. 2008;17:3919-28.
25. Lacbawan F, Solomon BD, Roessler E, El-Jaick K,
Domené S, Vélez JI, et al. Clinical spectrum of SIX3-associated
mutations in holoprosencephaly: correlation between genotype, phenotype
and function. J Med Genet. 2009;46:389-98.
26. Brown SA, Warburton D, Brown LY, Yu C-y, Roeder ER,
Stengel-Rutkowski S, et al. Holoprosencephaly due to mutations in
ZIC2, a homologue of Drosophila odd-paired. Nat Genet.
1998;20:180-3.
27. Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L,
Meinecke P, et al. Mutations in TGIF cause
holoprosen-cephaly and link NODAL signalling to human neural axis
determination. Nat Genet. 2000;25:205-8.
28. Roessler E, Muenke M. The molecular genetics of
holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:52-61.
29. Odent S, Le Marec B, Munnich A, Le Merrer M,
Bonaïti-Pellié C. Segregation analysis in nonsyndromic holoprosencephaly.
Am J Med Genet. 1998;77:139-43.
30. Ming JE, Muenke M. Multiple hits during early
embryonic development: digenic diseases and holoprosencephaly. Am J Hum
Genet. 2002;71:1017-32.
31. Dipple KM, McCabe ERB. Phenotypes of patients with
"simple" Mendelian disorders are complex traits: thresholds, modifiers,
and systems dynamics. Am J Hum Genet. 2000;66:1729-35.
32. Hahn JS, Plawner LL. Evaluation and management of
children with holoprosencephaly. Pediatr Neurol. 2004;31:79-88.
33. Joó GJ, Beke A, Papp C, Tóth-Pál E, Szigeti Z, Bán
Z, et al. Prenatal diagnosis, phenotypic and obstetric
characteristics of holoprosencephaly. Fetal Diagn Ther. 2005;20:161-6.
34. Wenghoefer M, Ettema AM, Sina F, Geipel A,
Kuijpers-Jagtman AM, Hansmann H, et al. Prenatal ultrasound
diagnosis in 51 cases of holoprosencephaly: craniofacial anatomy,
associated malformations, and genetics. Cleft Palate Craniofac J.
2010;47:15-21.
35. Mercier S, Dubourg C, Belleguic M, Pasquier L,
Loget P, Lucas J, et al. Genetic counseling and "molecular"
prenatal diagnosis of holoprosencephaly (HPE). Am J Med Genet C Semin Med
Genet. 2010;154C:191-6.
36. Barr Jr. M, Cohen Jr. MM. Holoprosencephaly
survival and performance. Am J Med Genet. 1999;89:116-20.
37. Levey EB, Stashinko E, Clegg NJ, Delgado MR.
Management of children with holoprosencephaly. Am J Med Genet C Semin Med
Genet. 2010;154C:183-90.
38. Roesler CP, Paterson SJ, Flax J, Hahn JS, Kovar C,
Stashinko EE, et al. Links between abnormal brain structure and
cognition in holoprosencephaly. Pediatr Neurol. 2006;35:387-94.
39. Plawner LL, Delgado MR, Miller VS, Levey EB,
Kinsman SL, Barkovich AJ, et al. Neuroanatomy of holoprosen-cephaly
as predictor of function: beyond the face predicting the brain. Neurology.
2002;59:1058-66.
40. Hahn JS, Hahn SM, Kammann H, Barkovich AJ, Clegg
NJ, Delgado MR, et al. Endocrine disorders associated with
holoprosencephaly. J Pediatr Endocr Met. 2005;18:935-41.
41. Hahn JS, Delgado MR, Clegg NJ, Sparagana SP, Gerace
KL, Barkovich AJ, et al. Electroencephalography in
holoprosencephaly: findings in children without epilepsy. Clin
Neurophysiol. 2003;114:1908-17.
42. Simon EM, Hevner RF, Pinter JD, Clegg NJ, Delgado
M, Kinsman SL, et al. The dorsal cyst in holoprosencephaly and the
role of the thalamus in its formation. Neuroradiology. 2001;43:787-91.
43. Agarwal SS. Medical genetics in India – what needs
to be done? Indian J Med Res. 2009;130:354-56.
44. Weaver DD, Solomon BD, Akin-Samson K, Kelley RI,
Muenke M. Cyclopia (synophthalmia) in Smith-Lemli-Opitz syndrome: first
reported case and consideration of mechanism. Am J Med Genet C Semin Med
Genet. 2010;154C:142-5.
45. Pineda-Alvarez DE, Dubourg C, David V, Roessler E,
Muenke M. Current recommendations for the molecular evaluation of newly
diagnosed holoprosencephaly patients. Am J Med Genet C Semin Med Genet.
2010;154C:93-101.
46. Sharp AJ. Emerging themes and new challenges in
defining the role of structural variation in human disease. Hum Mutat.
2009;30:135-44.
|
|
|
 |
|
