P
yoderma
gangrenosum (PG) is a rare, noninfectious neutrophilic dermatosis,
commonly associated with inflammatory bowel disease and rheumatoid
arthritis and also rarely with myelo-proliferative disorders and
hematological malignancies. The diagnosis of PG is clinical, along with
compatible histological findings. To our knowledge there is no report of
association of pyoderma gangrenosum with pure red cell aplasia in
children.
Case Report
A 4-year old boy, 1st born of non-consanguineous
parentage, who had normal developmental mile-stones, presented with
complaints of painful progressive skin lesions involving the buttocks and
both thighs since 4 months and restricted activity since 1 month. There
was history of fever accompanying the skin lesions. There was no similar
history in the past and no history of contact with tuberculosis. There
were no similar complaints in any other family members.
On examination, he was febrile, sick looking, had
pallor with generalized lymphadenopathy. His vital parameters were within
normal limits and anthropometry was normal. He had hepato-splenomegaly
(liver 4cm below right costal margin with liver span of 9cm, spleen 2cm
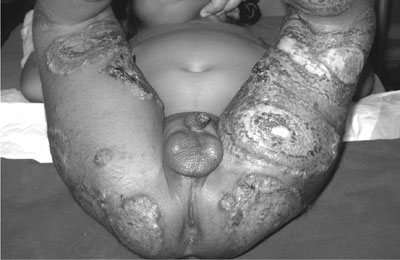
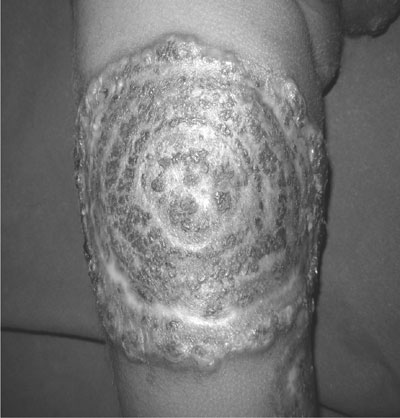
below left costal margin). Local examination showed multiple concentric,
targetoid lesions with ring ulcerations, crusting surface of margins
studded with vesicles, involving both thighs and buttocks (Fig.
1).
 |
|
(a) |
 |
|
(b)
Fig. 1 Cutaneous lesions – multiple
concentric, targetoid lesions showing extensive ulceration with a
ragged undermined edge and crusting surface of margins studded with
vesicles involving both thighs and buttocks. |
Investigations revealed a polymorphic leucocytosis
(total count 27900 cells/mm3; polymorphs
80%, lymphocytes 20%), microcytic hypochromic anemia (hemoglobin 7.3 g%),
and thrombocytosis (platelets 720000 cells/mm3). He had a raised ESR
(60/110 mm) and a high C-reactive protein (168.8 mg/L). His renal and
liver function tests were normal. Anti-nuclear antibody was negative.
Mantoux test (10 TU) was negative and HIV Elisa was non-reactive. His
immunoglobulin profile was also normal. Chest roentgenogram was normal and
ultrasonogram of abdomen revealed mild hepatomegaly. His blood culture did
not grow any organism. Bone marrow cytology revealed features of pure red
cell aplasia (striking reduction in the erythroid progenitors with a few
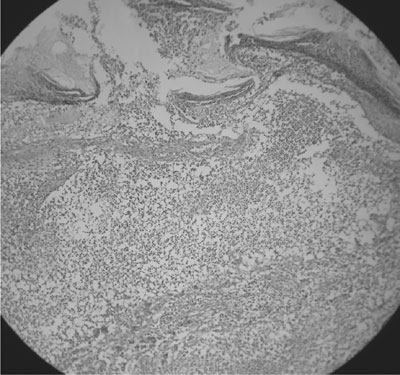
scattered normoblast), and the skin biopsy of the lesion (Fig.2)
showed focal ulceration with sub-acute inflammation and superficial dermal
abscess suggestive of superficial granulomatous pyoderma (abscess with
sheets of neutrophils and karyorrhectic debris with few multinucleate
giant cells and fibrinoid changes).
 |
|
Fig. 2 Skin lesion biopsy - ‘superficial
granulomatous pyoderma’ – abscess with sheets of neutrophils &
karyorrhectic debris with few multinucleate giant cells & fibrinoid
changes. |
With the above clinical picture and laboratory
findings, a final diagnosis of Pyoderma gangrenosum with pure red cell
aplasia was made and the boy was treated with intravenous antibiotics (amoxyclav)
for 1 week, and oral steroids (prednisolone 1mg/kg/day). He was also
transfused with packed red blood cells and started on iron and folic acid
supplementation. Fever settled in 4 days, with a dramatic healing of skin
lesions in 10 days. He became ambulant and was discharged on oral
prednisolone. Within a period of one month, the skin lesions had healed
significantly and he had normal hemoglobin (11g%). His spleen was not
palpable and the liver span had normalized. The steroids were tapered and
stopped. On a four-month follow up, the child is doing well with no
recurrences of the condition with normal hemoglobin levels.
Discussion
Pyoderma gangrenosum (PG) is defined as a destructive
necrotizing non-inflammatory ulceration of the skin clinically starting
with sterile pustules that rapidly progress and turn into painful ulcers
of variable depth and size with undermined violaceous borders(1). The
disease is more common in adults aged between 30 to 50 years and only less
than 4% of PG is seen in children(2,3).
Approximately 50-70% of PG is associated with an
underlying systemic disease(4), and the asso-ciated clinical conditions
commonly include inflammatory bowel disease, rheumatoid arthritis,
regional enteritis, chronic hepatitis, hematological disorders like
myelogenous leukemia, acute lymphoblastic leukemia, multiple myeloma,
poly-cythemia vera, myeloid metaplasia, pure red cell aplasia, and also
diabetes mellitus and immuno-deficiency(5). The lesions may precede, occur
simultaneously or follow the above clinical conditions and healing of
pyoderma gangrenosum lesions after successful control of the systemic
disease have also been reported(6).
There are 4 major clinical classification types of PG:
Ulcerative PG, Pustular PG, Bullous PG and the Vegetative PG(1). Our case
had chronic non-painful ulceration, absent violaceous border with slow
evolution - a form of superficial PG confined to the skin and known as
‘superficial granulomatous pyoderma’- a vegetative type of PG which is
usually not associated with systemic disease.
The diagnosis of PG is one of exclusion(6). The
management of this disorder begins with treatment of any underlying
disease and local or systemic glucocorticoids or immunomodulating
therapies(1,6). Management of skin lesions include (a) local wound
care, (b) topical therapy - topical steroids, cromolyn sodium, and
nicotine, and (c) systemic therapy - systemic steroids,
azathioprine, cyclosporine, tacrolimus, mycophenolate mofetil,
methotrexate, chlorambucil, thalidomide, colchicines, cyclo-phosphamide,
dapsone, minocycline, sulfapyridine, and TNF-µ inhibitors. Pulse
methylprednisolone, pulse cyclophosphamide, and intravenous immuno-globulin
have also shown benefit. Surgical treatment is useful only in extreme
conditions(1,6,7).
Our child presented with progressive skin lesions,
fever and pallor, and had no symptoms and signs of arthritis,
gastrointestinal disease and malignancy. The diagnosis was made based on
the skin biopsy and bone marrow cytology. The relation between pyoderma
gangrenosum and pure red cell aplasia is found to be rare in the
literature. Only two adult cases (a 72-year old man and an 80-year old
woman) have been reported so far with this rare association(8,9) and no
cases reported in the pediatric age group. This type of pure red cell
aplasia is often associated with neoplasia, mostly thymoma. It has been
reported earlier only in adults. The presence of serum inhibitors to
erythropoiesis and the response to steroid therapy suggests an autoimmune
phenomenon.
Contributors: SBS, KK, RR were involved in case
management. KK & JR were involved in review of literature and preparation
of manuscript. SBS will act as guarantor.
Funding: None
Competing interests: None stated.
References
1. Wollina U. Pyoderma gangrenosum – a review. Orphanet
J Rare Dis 2007; 2: 19.
2. Von den Driesch P. Pyoderma gangrenosum: a report of
44 cases with follow-up. Br J Dermatol 1997; 137: 1000-1005.
3. Graham JA, Hansen KK, Rabinowitz LG, Esterly NB.
Pyoderma gangrenosum in infants and children. Pediatr Dermatol 1994; 11:
10-17.
4. Powell FC, Schroeter AL, Su WP, Perry HO. Pyoderma
gangrenosum: a review of 86 patients. Q J Med 1985; 55: 173-186.
5. Crowson AN, Mihm MC Jr, Magro C. Pyoderma
gangrenosum: a review. J Cutan Pathol 2003; 30: 97-107.
6. Callen JP, Jackson JM. Pyoderma gangrenosum: an
update. Rheum Dis Clin North Am 2007; 33: 787-802.
7. Powell FC, O’Kane M. Management of pyoderma
gangrenosum. Dermatol Clin 2002; 20: 347-355.
8. Clayton R. Pyoderma gangrenosum with cellular
immunity defect and red cell aplasia. Proc R Soc Med 1977; 70: 571-572.
9. Wong CK, Pun KK, Lum CC, Lee SW, Ng MM, Wang CC.
Pyoderma gangrenosum associated with erythroid hypoplasia. Postgrad Med J
1990; 66: 312-313.


