|
|
Case Reports Indian Pediatrics 2008;45:505-507 |
||
|
Niemann-Pick Type C Disease |
||
|
Jayesh J Sheth
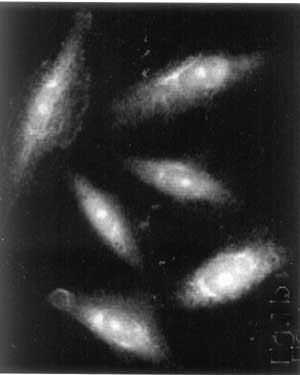
Key words: Niemann-Pick disease, India. Introduction Niemann-Pick disease type C (NPD-C) is an autosomal recessive panethnic neurovisceral lipid storage disorder with a wide spectrum of clinical phenotypes(1). The metabolic base of type ‘C’ remained enigma for a long time and is not yet fully elucidated inspite of a reclassification of the disease as a cellular lipid trafficking disorder. The prevalence in Western countries has been reported to be 1:1,20,000 to 1:1,50,000 live births(2). Initial manifestations can be hepatic, neurologic and psychiatric. Prolonged neonatal cholestatic icterus associated with progressive hepatosplenomegaly is the most common sign with spontaneous resolution by 2-4 months of age followed by rapid fatal liver failure in about 10% of cases without any neurological signs. In infants and young children, isolated hepatosplenomegaly may be the only sign of the disease. The most common presentation (about 60-70% of the cases) is late infantile and juvenile neurologic onset forms. Due to rarity and lack of investigational facility we report herewith a case of 4-year-old girl diagnosed with NPD type C. Case Report An Afghan girl was born at full term to a consanguineous muslim family with normal Apgar score. She was growing normally till one year of age. Later on, hepatomegaly with developmental delay was observed. Parents also noticed unexplained frequent falls without any sign of seizure. Proband was the 5th sib with one elder sister dying of respiratory failure and hepatosplenomegaly at the age of 5 years. Two elder brothers and one sister were clinically asymptomatic and growing normally till date. At the age of 4 yrs, she had neurological regression, hypotonia and facial dyskinesia. MRI study was suggestive of undermyelination. Bone-morrow examination revealed presence of storage cells suggestive for Gaucher or Niemann-pick disease and further confirmative diagnosis of lysosomal storage disorders was carried out. Lysosomal enzyme study from leucocytes was carried out for b-Glucosidase and Acid-sphingo-myelinase, which were 8.84 nmol/hr/mg protein (normal 8.21±3.11 nmol/hr/mg protein) and 1.16 nmol/hr/mg protein (normal 1.55±0.78 nmol/hr/mg protein), respectively. This was found to be normal making it unlikely for Gaucher or NPD type A or B. Further study was carried out on skin fibroblast for demonstration of unesterified cholesterol accumu-lation in cultured cells by filipin staining method, which is the characteristic of NPD type C(4). Intracellular accumulation of cholesterol was observed in skin fibroblast grown in presence of low-density lipoprotein confirming NPD type C in the proband (Fig. 1). Molecular study for mutation in NPC-1 or NPC-2 gene was not carried out due to lack of facility.
Discussion The clinical presentation of NPD type C is distinct from type A and B. It can be only hepatic manifestation like liver fibrosis, cholestasis, hepatomegaly and cirrhosis to neurological symptoms at two to 33 yrs of age(5-6). Hence precise diagnosis by demonstration of intravesicular cholesterol storage, deficient induction of cholesterol esterification and mutation study of NPC-1 and NPC-2 gene should be one of the confirmative approaches(4,7). Hepatic manifestation of NPD has been observed in Indian children but confirmative diagnostic test was not made(5). Present case is the first report from India where this was done. The phenotypic severity depends very much on the type of mutation. Those with NPC1 mutation have less severe phenotypes while HE1/NPC-2 gene mutation results in rapid onset of the phenotypes and death at 6 month to 4 month(7). The pattern of stored lipids varies in NPD-C, spleen and liver show moderate accumulation of unesterified cholesterol and spingomyeline resulting in hepatosplenomegaly(4). However, in brain tissue, neither cholesterol nor spingomyeline overtly accumulate but significant alteration of glycospingo-lipids have been reported. These alterations are not present in fetal tissues, but appear and gradually increase during the first two postnatal years. This explains the late development of neurological signs in the proband. Thus excess accumulation of glyco-spingolipids has been postulated as a causative of ectopic dendritogenesis in several lysosomal storage diseases including NPD-C(8). Recent studies have demonstrated an impaired trafficking of endogenously synthesized cholesterol(9). As a consequence free cholesterol accumulates in lysosomes and/or late endosomes, which is the most capricious specific marker for confirmation of NPD-C. Thus, NPD-C affects the transport of cholesterol through the late endosomal/lysosomal system and intracellular cholesterol homeostasis. This has formed the base of pharmacological trials to reduce the cellular influx of cholesterol or reduce the influx of less-complex glycolipids. Patterson, et al.(10) demonstrated that low cholesterol diet and varying combination of cholesterol lowering agents produce marked diminution in hepatic storage of unesterified cholesterol. Contributors: All authors were involved in literature search and drafting. Funding: ICMR. Competing interests: None stated.
| ||
|
References | ||
|
![]()