|
|
Case Report Indian Pediatrics 2004; 41:614-617 |
||||||||
Shubha R. Phadke
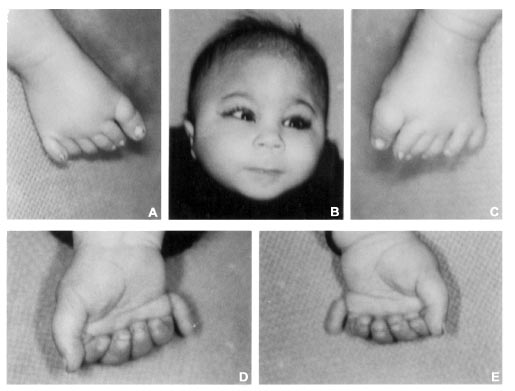
Abstract: We report a case of partial trisomy 13 with trigonocephaly, upslant eyes, long smooth philtrum, polydactyly, agenesis of right kidney and mild developmental delay. In this family phenotypically normal mother had pericentric inversion of chromosome 13 and the child (proband) received recombinant 13 from the mother. Genetic counselling of the family for reproductive risks and testing siblings of the mother for detection of balanced carriers is essential. Key words: Partial trisomy 13, C syndrome, trigonocephaly, polydactyly . The C syndrome also known as Opitz trigonocephaly syndrome is a multiple congenital anomaly/mental retardation (MCA/MR) syndrome. It was first reported by Opitz, et al. in 1969 and is characterized by trigonocephaly (prominent metopic sutures), multiple buccal frenula, polydactyly, skin laxity, primordial short stature, joint contractures and dislocations and various others physical findings(l-3). There may be mental retardation. Anomalies of internal organ involving heart, kidney and brain defects have been reported. Inheritance has been thought to be autosomal recessive but few cases with features similar to C syndrome have been reported with chromosomal abnormalities(4). Here, we report a case of partial trisomy 13 with features similar to C syndrome. This emphasizes the need for cytogenetic investigations of MCA/MR cases for providing accurate genetic counselling and prenatal diagnosis. Case Report Nine months old male child was referred for his unusual appearance, which was noted at birth. He was first-born child of a healthy non-consanguineous couple. Child was delivered at 36 weeks of gestation. Birth weight was 2kg and there was history of perinatal asphyxia and meconium aspiration. The child was kept in neonatal intensive care unit for 7 days. The family history was unremarkable. There was mild developmental delay. The infant was weighing 7 kg (3rd centile), occipitofrontal circumference was 44 cms (30th centile) and length was 66 cm (<3rd centile). He had prominent metopic suture (trigonocephaly), upslant of eyes, low set ears, long smooth philtrum, no dentition, noisy breathing, postaxial polydactly of all four limbs and a pilonidal sinus (Fig. 1). Systemic examination was within normal limits.
Ophthalmological examination showed no abnormalities. Ultrasonography of abdomen revealed non-visualization of right kidney with compensatory hypertrophy of the left. MRI brain detected no abnormalities and the skeletal survey was unremarkable. Cytogenetics The GTG banded karyotype from peri-pheral blood of the child was 46, XY, rec(13), dup(13) (qter ® q22 :: cen ® qter) (Fig. 2a). It means that there is extra material on the p arm of the chromosome 13. This extra segment is the part of q arm of chromosome 13 between the band q22 and the terminal end of q arm. It has resulted into the trisomy of the distal segment of 13 q between band q22 and the terminal end of the q arm. Mother’s karyotype revealed pericentric inversion of chromosome 13; 46, XX, inv(13) (p11q22) (Fig. 2b). The segment between the band p 11 and q22 has got inverted. Karyotype of the father was normal.
Discussion The present child had partial trisomy of chromosome 13. The clinical features present in this case are trigonocephaly, upslant of eyes, postaxial polydactyly, smooth and long philtrum, unilateral renal agenesis with developmental delay, which are similar to the features of ‘C’ syndrome. Cardiac defects and multiple alveolar frenula commonly seen in cases of ‘C’ syndrome were absent in present case(l). Some cases with phenotype similar to ‘C’ syndrome are associated with chromosomal abnormalitie(2-5). Chu, et al. have described a case of trigonocephaly (C) phenotype in a child with partial trisomy 13 and tetrasomy 13. The region q 22 ® q ter which is trisomic in this case is common to the trisomic cases reported by Chu et al(2). Trigonocephaly and polydactyly appear to be commonly seen features associated with trisomy of distal part of I3q(2,5,6). The syndrome of partial trisomy of 13q seems to have a characteristic phenotype. The features are trigonocephaly, polydactyly, renal anomalies, central nervous system anomalies and ear malformations, sharing similarity with ‘C’ syndrome. Trigonocephaly has also been reported with trisomy of part of 13q proximal to q21.1(7). Trigonocephaly which is the conspicuous feature of this case is also a feature of many other syndromes like Say-Meyer syndrome, Baller-Gerold syndrome, many chromosomal syndromes namely del (3q), del (7p), del (9p), del (11q). It is also found in association with various malformations of heart, brain, limbs, genitourinary tract but not conforming to any delineated syndrome. A large study of 237 cases of trigonocephaly revealed that in 184 cases, trigonocephaly was an isolated abnormality and these cases had normal mental function(7). In the study by Azimi, et al. also, 72% of trigonocephaly cases were non-syndromic and 5.6% of the cases were familial(8).In this family the phenotypically normal mother had pericentric inversion of chromosome 13, which resulted in recombinant 13 due to crossing over during meiosis. The mother being a carrier of balanced chromosomal rearrangement, the risk of recurrence of birth of a child with chromosomal imbalance is increased. Counselling for the risk of recurrence of malformed offspring and increased fetal loss is necessary once the carrier for such balanced chromosomal rearrangement is identified. Though the risk varies depending on the chromosome involved, length of the inverted segment etc; the general empiric risk of birth of a malformed child is 5-10%(9). Data from previously reported inversion with similar breakpoints (p11, q21/22) shows empiric risk of recurrence of unbalanced offspring to be 7.5% for female heterozygotes(10). The risk of preterm losses increases to 27%(10). Approximately 50% of phenotypically normal offspring will be heterozygotes, subject to similar risks and perpetuation of inversion to future generation. Karyotyping of the parents and the siblings of inversion carrier is indicated to identify carrier and offer them genetic counselling and prenatal diagnosis. Prenatal cytogenetic analysis from chorionic villi or amniotic fluid can provide diagnosis and help the family to avoid the recurrence. Contributors: SRP made the clinical and cytogenetic diagnosis of the case. SJP did review of literature. Both the authors contributed to writing of the paper. Funding: Nil. Competing interests: None stated.
| ||||||||
|
![]()