|
|
Brief Reports Indian Pediatrics 2001; 38: 658-662 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Langerhans Cell Histiocytosis Involving Single System |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
From the Regional Cancer Center, Post Box No.
2417, Medical College, P.O. Thiruvananthapuram, Kerala 695 011,
India. Manuscript received: May 26, 2000; Initial review completed: June 25, 2000; Revision accepted: December 21, 2000. Langerhans cell histiocytosis (LCH) is an uncommon disease characterized by the idiopathic proliferation of Langerhans cells or their marrow precursors. The term embraces the whole clinical spectrum of the disorder from single bone lesions to an aggressive widespread multisystem disease. The cause of LCH is not firmly established and most investigators in the field have long suspected that LCH is immunologic disorder either in its etiology or in its pathophysiology(1,2). Recent evidence suggest that LCH is a clonal disorder rather than reactive disease(3). LCH is classified according to sites of involvement into single system disease and multisystem disease(4). Single system disease can be either unifocal or multifocal. Mutlisystem disease can be either without organ dysfunction or with organ dysfunction. Clinical course of LCH with single system disease is usually benign with high chance of survival(5,6). This study evaluated the clinical profile, treatment methods and outcome of children with LCH involving single system. Subjects and Methods All children with a diagnosis of LCH registered in our center during January 1984 to 1996 were analyzed retrospectively. Thirty seven were found to have LCH of single system and they were the subject of this study. In all these children diagnosis was based on clinical and histopathological data. Extent of disease at diagnosis was assessed by the following examinations in all patients. These included a complete medical history, physical examination, laboratory and radiologic eva-luation, urinalysis, liver function tests, renal function tests and skeletal survey. Bone marrow aspirations and biopsies were done when peripheral counts were abnormal. Additional investigations were done in selected cases. The extent of disease was documented as per the recommendation of Histiocyte Society(4). Patients with single system disease were stratified as follows: (a) Unifocal: single bone lesions, isolated skin disease, solitary lymphnode; and (b) Multi-focal: Multiple bone lesions, multiple lymph-node involvement. Treatment consisted of surgical inter-vention (biopsy, curettage or excision) radio-therapy, chemotherapy or combination of modalities. Patients with localized disease were treated with surgery with or with- out radiotherapy. Chemotherapy with Inj. Vinblastine, Prednisolone with or without Cyclophosphamide for 6-12 months was used in patients with multiple lesions at diagnosis. Treatment response was assessed as per recommendation of Histiocyte Society - as better, intermediate and worse. The duration of survival was calculated from the date of diagnosis. Survival probability was calculated by Kaplan Meier Method. Results Thirty seven children were found to have disease affecting single system. Their ages ranged from 10 months to 14 years (median - 5 years). The Male : Female ratio was 2 : 1. The median follow up of patients was 66 months. Of the 23 children with unifocal disease, 21 had involvement of skeletal system and 2 had isolated cervical lymphnode involvement. There was no case of isolated skin disease in this series. Skull bones were affected in almost all patients (20/21) except one who had isolated lesion of femoral bone. Scalp bones were affected in 14 children, mandible and orbital bones in 2 children each and greater wing of sphenoid and mastoid in one each. Table I shows the pattern of disese, treatment modality and current status of 14 children with multifocal disease. Of these 14 children, disease affecting the skeletal system was seen in 13 and one child had multifocal involvement of lymph nodes. Skull bones were involved in 10 children; femur in 4; ileum, vertebrate and clavicle in 2 children each and humerus, radius and maxilla in one each. The patient with multifocal involvement of lymph nodes had generalized lymph-adenopathy. Thirty six patients were evaluable for treatment response. One patient with multi-focal disease did not receive any treatment because of parental refusal. Sixteen patients with unifocal disease received biopsy/curett-age as the only form of treatment. Radio-therapy to local area was given to 7 patients. The primary sites were skull-1, mastoid-1, mandible-1, femoral neck-1, orbit-2, cervical lymphnode-1; none of them received any systemic form of treatment. Of the evaluable 13 patients wth multifocal disease, 2 patients had biopsy only. Radiotherapy was given to 7 patients, chemotherapy for 3 and one child received both chemotherapy and radio-therapy. Better responce was seen in all patients except two who had intermediate response. Recurrence was noted in 4 patients with unifocal disease (17.3%). One patient who had disease in the greater wing of sphenoid developed diabetes insipidus (DI) and recur-rence in skull and ileum at 26 months. Recurrences were confined to the skeletal system alone in other 3 patients. Four of 13 patients (30.5%) with multifocal disease developed recurrences, one had recurrence in gum, one had recurrence in skull and third patient had recurrence in gum and skull. The patient with involvement of lymph nodes developed recurrence in the lymph nodes itself. All these recurrences were seen within 30 months of initial diagnosis. One patient in each group had multiple recurrences. Of these 36 patients who were on follow up, one patient who developed DI opted for treatment elsewhere and one patient was lost for follow up. One patient who had involvement of multiple lymph nodes died due to pneumonia at 65 months. Thus overall survival in this group was 94.4% and event free survival was 77.1%. Table I - Summary of 14 cases with single system multiple site involvement
RT - Radiotherapy, V - Vinblastine 6 mg/m2 IV, Prednisolone 40 mg/m2 PO daily, m - months, Fr - Fractions, cyclophosphamide - 50 mg/m2/wk. MTX - T. Methotrexate 20 mg/m2/week, 6MP-T, Purinethol 50 mg/m2 PO daily, VCR - Inj. Vincristine 1.5 mg/m2, LFU - Lost for follow up, NED - No evidence of disease.
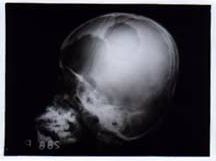
Discussion LCH is a rare disease with highly variable clinical presentation. This study evaluated the disease course and outcome of Pediatric LCH with single system involvement. About 50% of our patients presented with single system disease of which 92% had involvement of skeletal system. Bone involvement is very common in LCH but the reported incidence of isolated skeletal disease has not been uniform. In the large French series, 31% presented with isolated uni or bifocal bone lesions and 19% had multifocal bone involvement(7). Bone involvement with or without other affected sites has been observed in 80 to 100% of LCH(7,9). Lymph node involvement is seen in a small number of patients, found in fewer than 10% of children at presentation. Lymph node involvement was seen in 8% of our patients. Skin is reported as the only affected site in about 10 to 13% of cases(9,10). None of our patients had isolated skin disease. The clinical course of LCH in patients with single system disease is generally benign with high chance of spontaneous remission and favorable outcome(5,6). Bony lesions require simple curettage or even a liberal biopsy. Infiltration of affected site with steroids recently has also been reported to be an effective and safe modality(11), Radiotherapy is reserved for disease that is inaccessible to intra-lesional injection of steroid or that is potentially compromising vital structures (e.g., optic nerve, spinal cord). In such case, a short course of radiation (450 cgy to 600 cgy) may prove effective. When multiple bones are affected, treatment generally consists of systemic agents. When treatment of one site is clinically necessary, surgical removal, local injection of steroids or irradiation may be considered and treatment with potentially toxic systemic agents may be avoided. For isolated lymph node involve-ment or isolated nodular lesion of skin, surgical excision is the therapy of choice. Disseminated skin lesions may respond to topical application of steroid or nitrogen mustard(12). Radiotherapy was given to 14 patients in our series. Radiotherapy to skull was given to patient when the skull lesions were large (Fig. 1). Cervical lymphadeno-pathy was massive in one patient and excision was not possible and radiotherapy was considered. Radiotherapy was given to other patients considering the site and size of the disease. Two patients received chemotherapy with Inj. Vinblastine and Prednisolone for 6 months and one patient received oral Cyclophosphamide in addition for 12 months. Recurrence of the disease was observed in 8 patients, 4 in the unifocal group (17.3%) and 4 in the multifocal group (30%). First recurrence was noted within 30 months of initial diagnosis. All the 3 patients who received chemotherapy after initial diagnosis developed recurrences. Hence the effective-ness of chemotherapy in preventing late disease in this group of patients is questionable. There is a significant difference in the overall survival and disease free survival. This is because recurrences, even if they are multiple do not lead to mortality. Even though, there is a suggestion that LCH is clonal disorder. Its behavior is different from that of a classical malignancy. Late deaths were reported in few studies where long term follow up was done, indicating the chronicity of disease. One death occurred in our study at 65 months due to pneumonia. Autopsy was not done in that case and hence we can’t rule out the possibility of reactivation of the disease. In conclusion, although overall survival is good, recurrences are seen even in patients with single system disease at diagnosis. The role of chemotherapy in preventing these recurrence is questionable.
Funding: None.
Fig. 1. Two large lytic lesions in the skull.
|
![]()