|
|
Review Indian Pediatrics 2001; 38: 631-639 |
|||
|
Carrier Detection and Prenatal Diagnosis in Duchenne/Becker Muscular Dystrophy |
|||
From
the Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute
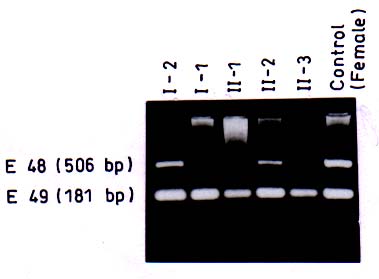
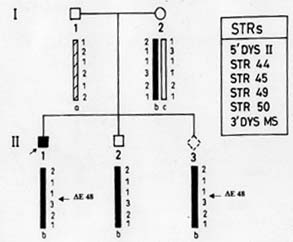
of Medical Scien-ces, Raebareli Road, Lucknow 226 014, India. Majority of genetic disorders place a considerable burden on the families per-petuating the condition for the lack of effective treatment. Duchenne/Becker muscu-lar dystrophies (D/BMD) are such lethal disorders caused by mutations in the dystro-phin gene. DMD is a common disease affect-ing 1/3500 male births, while BMD is milder and less frequent. It presents with muscular weakness, hypertrophy of the calf muscles, positive Gower’s sign, and Pradhan’s valley sign(1). Due to X-linked nature of the disorder , males carrying the mutated gene are affected, while females become carriers of the disease. Diagnosis of patients with D/BMD is usually definitive based on clinical, patho-logical and biochemical findings, although it is increasingly being confirmed by molecular analysis. Due to the lack of efficient rehabilitation and treatment of progressive muscular dys-trophy, counseling and prenatal diagnosis are options that medical geneticists can offer today, and their decision depends on infor-mation of the carrier status. Practically, if the mother of an affected boy (proband) has another affected relative, she is an obligate carrier. If there is an affected brother or one affected son, she is a possible carrier. But in most families there is only one affected patient. Therefore, female relatives of affected males are candidates for carrier assessment. However, the ascertainment of carrier status is one of the basic dilemmas in genetic counseling in X-linked recessive disorders because female carriers are usually asymptomatic. Several biochemical and mole-cular methods have been suggested to solve this dilemma. Among the biochemical methods, the serum creatine phosphokinase (CPK) level has been most frequently used for carrier detection in DMD. However, the CPK level is informative in only 70% of the female carriers(2,3). About one-third of the female carriers show myopathic changes in muscle histology and abnormalities have also been found histochemically and by electron micro-socopy(4). Immunohistochemical methods using antibodies against dystrophin protein have also been used for carrier detection(5,6) Most of these methods, however, are either invasive or not specific for D/BMD and there-fore cannot be used for definitive ascertain-ment of status. Recent advances in molecular techniques have made it possible to detect carriers and provide prenatal diagnosis with almost 95% confidence (high specificity). The dystrophin gene is about 2.4 Mb in size, consisting of 79 exons and is localized at Xp21. Most of the mutations in the gene are seen in two hotspots located at proximal and central regions of the gene called the ‘deletional hotspots’(7,8). About 65-70% of the patients show intragenic deletions of one or several exons of the gene which can be detected by multiplex PCR (Polymerase chain reaction) or Southern blotting(9,10) using DNA from patient’s blood. However, carrier analysis by molecular methods in females is still difficult due to the presence of a normal copy of the gene in the heterozygous carrier females (2 X-chromo-somes). In these cases quantitative PCR or quantitative Southern blotting have to be carried out and the products are assessed by densitometry (11-13). The process is how-ever, cumbersome and requires strictly controlled conditions. Moreover, in 30% of the cases of D/BMD, no gross deletions are detected in the dystrophin gene due to the presence of point mutations. Various point mutation screening protocols such as SSCP (Single Strand Conformation Polymor-phisms), HA (Heteroduplex Analysis), CMC (Chemical Mismatch Cleavage), etc. have been designed but the success rate is limited due to large size of the gene(14-16). An alternative approach for carrier detection is by DNA based linkage analysis using highly polymorphic intragenic short tandem repeat (STR) markers(17). It has 95% accuracy due to the possibility of recombina-tion. The above method depends on the segregation of the X-chromosomes from mother to the affected and unaffected offsprings. Several STR markers for the gene are used for the method (http//www.dmd.nl). Linkage analysis is suitable both for deletional as well as non-deletional cases of D/BMD, which allows proper carrier detection and prenatal analysis. Various molecular methods as described above are available for prenatal diagnosis and carrier analysis. The details of these methods are as follows: (1) Gene deletions by Multiplex-PCR The rapid detection of deletions by PCR allows proper counseling in the new families. Specific DNA regions are amplified in a multiplex reaction using Chamberlain’s(9) and Beggs’(18) and some additional primers. Usually the laboratories use 25 pairs of primers, which are able to detect about 98% of the deletions. The deletion can be identified from the pattern of the bands as visualized on ethidium-bromide stained agarose gels. Once the deletion has been detected, it becomes a marker for the family and prenatal diagnosis can easily be provided. For example, in Fig. 1 the proband of a DMD family had shown the deletion of exon 48 and this deletion was used as a direct marker for prenatal diagnosis in the family. The above method, however, has limitations, as it can not be used for determination of carrier status in females because of heterozygosity.
(2) Gene deletions by Southern blotting In this method the DNA is digested by restriction enzymes which recognize and cut at specific sites. The digested DNA is subjected to electrophoresis and hybridized to complimentary cDNA probes. The labeling of probes by radioactive nucleotides allows detection by autoradiography(10). The proce-dure is however, tedious and is especially used when single exon deletion is detected by PCR analysis. This is because single exon deletion may result from failure of amplifica-tion of that exon due to polymorphism. Among affected males, 5-10% show duplica-tions of one or more exons of the gene, which can also be detected by Southern blotting. (3) Quantitative and real time PCR These are further refinements of the conventional PCR technology. However, these are mostly in the investigational stage and require stringent conditions. The sample has to be analyzed in the exponential phase of the PCR amplification before the plateau phase is reached for proper quantitation. In real time, the progress of PCR can be monitored and accordingly the reaction can be terminated at the desired point(19, 20). The inherent complexities of these methods and high costs preclude their use for routine diagnosis.
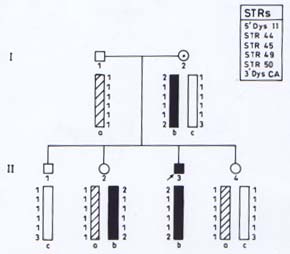
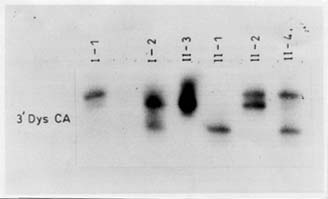
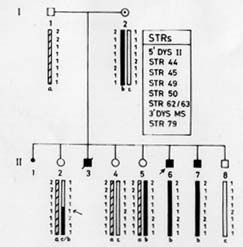
Microsatellites are short tandemly repeated sequences(21), which have been identified as byproducts of the Human Genome Project. There are approximately 50,000-100,000 (CA)n loci (a subclass of STRs) in the human genome. These frequently polymorphic loci have been exploited in carrier detection of many genetic diseases by linkage analysis. DNA based linkage analysis, using intragenic (CA) repeat markers of dystrophin gene has been found to be a powerful approach for carrier detection of non-deletional as well as deletional D/BMD families. Due to large size of the gene, intragenic meiotic recombinations are also possible. Therefore, several intragenic and flanking STRs of dystrophin locus are being studied to provide accurate carrier status and prenatal diagnosis. The method requires the sample of proband in addition to other family members. The informativeness of the markers depends on the level of heterozygosity in the population. Hence, the markers selected have to be specific for the target population. Usually 4-6 STR markers for the deletion prone regions of dystrophin gene are used and the haplotype of the specific X-chromosome is deduced. The PCR products are run on polyacrylamide gel and visualized by autoradiography(17). Determination of Carrier Status of Female Relatives The usefulness of (CA)n markers in the identification of carrier status of female relatives and for prenatal diagnosis in affected families is being explained with the help of some examples from our laboratory records. In family 1 (Fig 2A), the mother was an obligate carrier due to family history of the disease. The proband had not shown any deletion. Therefore, linkage analysis was directly carried out to know the status of two female sibs who were at risk of being carriers. Six (CA) n repeat markers of the dystrophin gene (5’DYS-II, STRs 44, 45, 49, 50 and 3’DYS-MS) were amplified by PCR using DNA samples from all the family members. The products were separated by gel electro-phoresis and assessed by autoradiography. The mother (I-2) was found to be hetero-zygous for two STRs, 5’DYS II (2,1) and 3’CA (2,3) loci (Fig. 2A). The haplotype ‘b’ of the mother inherited by the proband was linked with the disease while ‘c’ is normal. By following the segregation of ‘b’ and ‘c’ in the offsprings we can determine the status of females in the family. It was observed that proband (II-3) and female sib (II-2) inherited the maternal haplotype ‘b’ while female sib (II-4) inherited the haplotype ‘c’, thereby suggesting that (II-2) was a carrier while (II-4) was a non-carrier. Figure 2B shows the electrophoretic pattern of 3’DYS CA loci in all the family members. In family 2 (Fig.3) there was a history of the disease and the proband had not shown any intragenic deletion. Carrier status of three female siblings in the family was determined using eight (CA)n repeat markers. By haplotype analysis, it was found that mother was informative (heterozygous) for three (CA) loci, STRs 5’ Dys II (2,1), 50 (1,2) and 62/63 (2,I). The feamle sib (II-5) was suggestive as a carrier as she shared the haplotype ‘b’, which was inherited by affected brothers. Another female sib (II-4) was ascertained as non-carrier. The carrier status of (II-2) could not be ascertained as she inherited a mixed maternal haplotype (b/c) suggesting the occurrence of recombination between STRs 49 and 50 (Fig. 3). Therefore, carrier analysis can not be predicted in 2-5% of cases where intragenic recombination has taken place. In family 3 (Fig. 4), one son (II-1) was clinically diagnosed as DMD at 6-years of age and he showed deletion of exon 48 (Fig.1). This deletion was used as a marker for prenatal diagnosis in this family and the same was found to be present in the CVS (II-3). The CPK level of proband was found to be raised while mother’s CPK level was in the normal range. No intragenic deletion was observed in the unaffected male sib (II-2). By haplotype analysis, the mother (I-2) was found to be informative for three STRs, 5’DYS-II (2,1) 44 (1,3), and 49 (Fig.4). It was observed that proband (II-1), unaffected brother (II-2) and fetus (II-3) inherited the same maternal haplotype ‘b’. However, no deletion was detected in II-2 and was clinically normal. This suggested that the mother was a germline mosaic. This can be explained on the basis that mother had both normal and mutated germ cells. The proband (II-1) and CVS (II-3) had inherited the mutated germ cells while II-2 had inherited the normal germs cells from the mother. In D/BMD, about 5-10% of cases has been attributed to germline mosaicism(22). It is therefore important to carry out prenatal diagnosis in all future preganancies if there is one affected child in the family. Since the fetus in the above family was found to be an affected male, the parents were counseled accordingly.
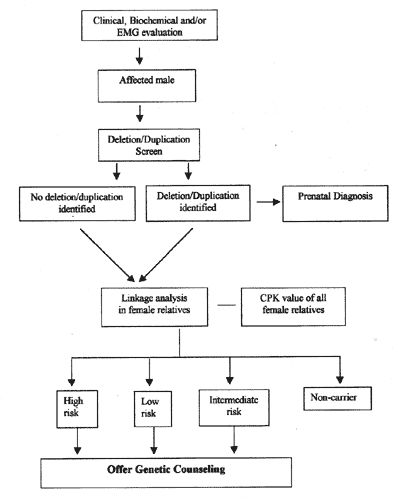
(5) Other Techniques The FISH (Fluorescent in-situ hybridi-zation)(23) technique can be employed for carrier detection in female relatives in deletional D/BMD cases only. However, its use is limited due to high cost of the fluorescent probes and equipment required. Single strand conformation polymorphism (SSCP) analysis(14), heteroduplex analy-sis(15), RT-PCR (Reverse transcription PCR) (24) and protein truncation tests (PRI)(25-26) are used mainly for point mutation detection. But these techniques are not feasible for routine diagnostic purposes. Several centers in India apart from our center at Sanjay Gandhi PGIMS provide the molecular diagnosis for D/BMD. Our laboratory is involved in mutational analysis, carrier detection, and prenatal diagnosis in D/BMD families for the last 10 years(27-29). Till date, we have analyzed around 327 D/BMD patients. Out of these, 220 (67.3%) were found to have intragenic deletions and 107 (32.7%) were nondeletional cases. Carrier detection in the female relatives has been carried out in about 25 D/BMD families. In our study, 58-70% informative-ness was observed for central region STRs and 27-47% for 5’/3’ region STRs. Prenatal diagnosis was successfully provided to 5 families. The initial screening involves deletion identification by multiplex-PCR analysis/Southern hybridization. Linkage analysis is being used in both deletional and non-deletional families for carrier detection. The approach to molecular diagnosis for Duchenne muscular dystrophy has been summarized in Fig. 5.
Fig. 5. Flow chart suggesting the approach for carrier detection and prenatal diagnosis in D/BMD. Acknowledgements We are grateful to Dr. L.S. Chaturvedi, Dr. Sandhya Srivastav and Dr. Monisha Mukherjee for the molecular analysis and help in preparing the paper. We are also grateful to our consultants, Dr. S.R. Phadke and Dr. S. Pradhan. Contributors: IP designed the review, analyzed the data and drafted the paper. BM supervised the work, made finer corrections and will act as guarantor for the paper. Funding: Council for Scientific and Industrial Research and Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow. Competing interests: None stated.
|
![]()