|
|
Case Reports Indian Pediatrics 2000;37: 670-673 |
||||||
|
Glycogen Storage Disease Type III |
||||||
|
Type III glycogen storage disease (GSD), an autosomal recessive disease, is caused by deficient glycogen debranching enzyme (GDE) activity. Patients with GSD are usually diagnosed in infancy or early childhood with hypoglycemia, hepatomegaly, poor physical growth, and a deranged biochemical profile(1). We report a case of GSD type III with predominantly liver involvement.
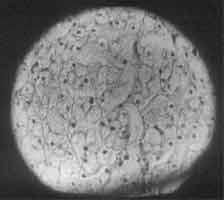
A 2½-year-old male child born of consanguineous marriage residing at Rajasthan was referred to our center for progressive abdominal distension, hepatome-galy and failure to thrive for the last 1 year. During the first year of life he had 2-3 attacks of afebrile seizures and was investigated at Rajasthan, his CT scan and EEG were normal and he was put on phenobarbitone since then. On examination he had round face, his weight was 11 kg and height was 80 cm, both were less than 3rd percentile for his age. Icterus was not present but he had hepatomegaly of 15 cm and splenomegaly of 4 cm, with no fluid in the abdomen. Skeletal hypertrophy was not present and on slit lamp examination KF ring was not seen. Laboratory test results were as follows: normal hemogram, total protein 7 g/dl, serum albumin 4.4 g/dl, alkaline phosphatase 335 U/L, serum bilirubin 2 mg/dl, SGOT 587 U/L, SGPT 361 U/L, normal coagulation profile, fasting blood sugar 29 mg/dl, post prandial blood sugar 108 mg/dl, serum cholesterol 495 mg/dl, serum triglyceride 372 mg/dl, blood pyruvate 3.8 mg/dl, blood lactate 1 mM and normal hepatitis B and C serology. Glucagon challenge test after overnight fast resulted in blood sugar values: fasting blood sugar–39 mg/dl; 30 min–36 mg/dl and 60 min–41 mg/dl. After meals, blood sugar was 84 mg/dl and 1 hour after glucagon administration blood sugar increased to 104 mg/dl. Erythro-cyte glycogen content was 417 mcgm/g Hb (normal value < 150) and amylo-1, 6-glucosi-dase activity in the leucocytes was 0.04 nanomoles of glucose/mn X mg protein. The child was suspected to have glycogen storage disorder based on findings of gradual abdominal distension, history of convulsions, massive hepatomegaly, hypoglycemia, hypertriglyceridemia, hypercholesterolemia and abnormal liver enzymes. He was diagnosed GSD type III based on liver histology and enzyme analysis. Liver histology with hematoxylin-eosin stained reaction showed hepatocytes with vacuolated cytoplasm and central nuclei (Fig. 1). No fibrosis or inflammatory cell infiltration seen. As the tissue was fixed in formalin PAS stain did not show PAS positive material in the cytoplasm. Increased glycogen content in his erythrocytes and deficiency of amylo-1-6 glucosidase activity in the leukocytes was in agreement with a diagnosis of glycogenosis type III.
GSD type III is a metabolic disease in which glycogen with very short outer arms, the so called limit dextrin, is stored in liver or muscle because of a deficiency of debranching enzyme(2). Approximately 80% of patients are GDE deficient in both liver and muscle (type IIIa) and 15% of patients have GDE absent in liver but retained in muscle (type IIIb). It is an autosomal recessive disease, presents in early childhood and has an incidence of 1:50,000 to 1:70,000(2). Our case also presented in early childhood. Liver, skeletal muscle and heart are the main organs involved in various levels of severity. In our case there was only hepatic involvement. Gross hepatomegaly, hypogly-cemia, seizures and growth retardation are the main presentations. Our case also had similar presentation. Majority of the cases with hepatic involvement present with protruded abdomen because of gross hepatomegaly and also have raised serum aminases and serum cholesterol. During childhood and early adulthood the symptoms seems to regress and normal adulthood appears in most patients except few cases who may develop cirrhosis with or without sequelae of portal hypertension(3). Therefore hepatological follow up is required during adulthood. Hepatomegaly gradually resolves concurrent with decrease in elevated transaminase level. In our case also liver size has decreased from 15 cm to 6 cm with decrease in serum transaminase levels. The serum concentration of uric acid, lactate and ketones were normal. In the present case the glucagon test did not show a significant rise in fasting blood sugar. However, the rise in post prandial blood sugar was significant. Differ-entiation between type I and type III GSD was made by the intravenous glucagon test in the immediate post-prandial period when there is usually a rise in blood sugar levels in type III but not in type I patients(4). Deficiency of amylo-1-6 glucosidase can be demonstrated in one of the following tissue; leukocytes, erythrocytes, liver, muscle, fibroblast or chorionic villi(5). In our patient we did erythrocyte glycogen content as well as amylo-1-6 glucosidase activity in leukocyte in France as these assays are not available in India. It revealed increased glycogen content and deficency of amylo-1-6 glucosidase activity in the erythrocytes, that confirms the diagnosis of glycogenosis type III. The patient’s father’s and maternal grandmother’s amylo-1-6 glucosidase activity in the erythrocytes did not fit with their heterozygous status. The advocated treatment for GSD type III comprises frequent high protein feedings during the day and a high protein snack at night; energy is distributed as 45% carbohydrates, 25% protein and 30% fat(1). Carbohydrates should be given frequently round the clock when the patient is young. Gastric drip feeding at night may be introduced in the infant if hypoglycemia is a problem(6). Uncooked corn starch therapy can be started in the older child and is very useful(7). Our patient was explained to take every two hours feeds of corn starch, rice and lentils in between the meals. He was followed up regularly for height, weight, liver size and serum transaminase assays. He has gained height and weight, his liver size has regressed and serum transaminase levels have come down remarkably. SGOT was 189 U/L and SGPT was 259 U/L after nine months of diet therapy. The prognosis for a relatively normal life is good. Prenatal diagnosis and carrier detection is possible with the help of recent identification of the highly informative DNA polymorphic marker in the AGL gene(8).
|
![]()