 |
|
Case Reports Indian Pediatrics 2000;37: 659-662 |
|||||||||||||
|
Idiopathic Cranio-osteoarthropathy |
|||||||||||||
|
Sushil Kr. Kabra Manju Ghosh A.K. Gupta* P.S.N. Menon
Idiopathic hypertrophic osteoarthropathy (HOA) is a rare entity and accounts for less than 5% of all cases of HOA(1). The classical form known as Pachydermoperiostitis or primary idiopathic osteoarthropathy is characterized by coarsening of facial features with thickening and furrowing of the face, clubbing of the digits with periosteal new bone formation. Infants with a distinct variety of HOA associated with cranial enlargement, delayed closure of cranial sutures and skin manifestations have been reported(2). A family with three affected sibs having cranial suture defects and idiopathic HOA was described(3). We report a case of idiopathic cranio-osteoarthropathy without skin manifestations in an Indian child.
A 2½-year-old boy presented with deformities of extremities and cranium since birth. He was born to a 17 year old mother married nonconsanguineously. The father was 20 years old. Parents were normal. The second child born after him was a preterm delivery and died after one day but he did not have any abnormalities at birth. The child had normal developmental milestones apart from mild motor delay. He weighed 9.5 kg (< 5th centile and his height was 83 cm (< 5 th centile) with an upper lower segment ratio of 1.2 : 1. The head circumference was 45 cm (< 5 the centile). He had a widely open anterior fontanel and frontal bossing. Facies were normal except a wide nasal bridge. He had drumstick clubbing of fingers and toes (Fig. 1). The long bones were cylindrical and thick and tibia was bowed. There was restriction of supination and pronation at elbows and all movements at knee joint. Examination of other systems revealed no abnormality. There was no evidence of arthritis or effusions in any joints. There were no skin lesions. His hemogram, renal function tests, liver functions, calcium, phosphate and alkaline phosphatase were normal. Skeletal survey revealed widening of medullary spaces of long bones, periosteal and endosteal reaction in middle two thirds but the ends of long bones were normal (Fig. 2). X-rays of hands and feet showed medullary thickening and acro-osteolysis (Fig. 3). Based on these findigns a diagnosis of cranio-osteoarthropathy was made.
There are various case reports of HOP with associated skin changes like pachyderma (Pachydermoperiostosis), thickening of skin and excessive sweating(4-6). Pachydermo-periostosis in a thirteen year old Indian boy presenting as acromegaly has been reported(7). Two reports of familial cases with crani-osteoarthropathy(2,3) and few sporadic cases without skin changes(1,8,9,10) have been described. Another Indian boy with idiopathic hypertrophic osteoarthropathy without skin manifestations and cranial changes has been reported(11). Our case had characteristic findings in the form of drumstick clubbing of fingers and toes, bowing of long bones and widely open cranial structures. These findings were noticed at birth. He did not have any evidence of joint effusions, pain or skin manifestations. All causes of secondary HOA were excluded. Other differential diagnoses include conditions like congenital syphilis, idiopathic hyperostosis, Caffey’s disease, Engelman syndrome and hypervitaminosis A. These conditions have characteristic radiological and clinical findings. Though all these conditions may be associated with periosteal reaction, hyperostosis and new bone formation, none of them show characteristic clubbing as seen in cranio-osteoarthropathy. Pathogenesis of this rare entity is not clear. Sluggish blood flow in pachydermoperiostosis as opposed to increased flow in pulmonary osteoarthropathy has been suggested based on angio-graphic studies(12). Occurrence of two or three cases in one generation suggests autosomal recessive inheritance(3). Autosomal dominant pattern of inheritance has also been described in pachydermoperiostosis(12). Recently, association between primary HOA and the major histocompatibility genes has been studied in 4 mexican families. All four families shared HLA-DRY, SC31 segment suggesting involvement of this haplotype in pathogenesis of primary HOA(13). The disorder is self limiting as cranial stuture widening and subperiosteal new bone formation decreases with age but clubbing and arthritis persist. This may suggest effect of some endogenous or exogenous factors as pointed out earlier(2). The age of presentation is variable. Our case and one of the cases described by Regeneto et al.(3) presented at birth while others were diagnosed in early infancy. Parents should be counselled about self limiting nature of the disease. Possibility of prenatal diagnosis should be investigated by ultrasonography in second trimester.
|
![]()
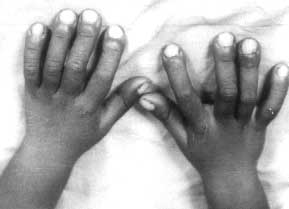
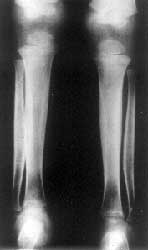
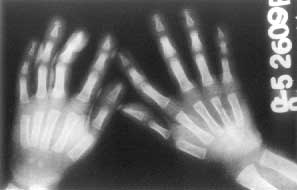 Fig. 3. X-ray hands showing medullary thickening
Fig. 3. X-ray hands showing medullary thickening