|
|
Review Articles Indian Pediatrics 2000;37: 615-625 |
||||||||||||||||||||||||||||||||||||||||
|
Recent Advances in Chromosome Breakage Syndromes and Their Diagnosis |
||||||||||||||||||||||||||||||||||||||||
|
From the Genetics Unit, Department of
Pediatrics, All India Institute of Medical Sciences, Reprint requests: Dr. Roli Mathur, Genetics
Unit, Department of Pediatrics, Manuscript received: July 2, 1999; Initial
review completed: Revision accepted: December 24, 1999. Chromosome instability is a characteristic cytogenetic feature of a number of genetically determined disorders collectively called as the chromosome breakage syndromes or DNA-repair disorders. They are characterized by susceptibility to chromosomal breakages, increased frequency of breaks and interchanges occurring either spontaneously or following exposure to various DNA-damaging agents. These diseases are a group of genetic disorders sharing a number of features. They are all autosomal recessive, show an increased tendency for chromosomal aberrations and to develop malignancies. The principal diseases in this group having a diverse etiology and clinical manifestations include Fanconi anemia (FA), ataxia telangiectasia (AT), Nijmegen breakage syndrome (NBS), Bloom syndrome (BS), xeroderma pigementosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD). The underlying defect in these syndromes is the inability to repair a particular type of DNA damage. A number of repair disorder phenotypes are caused by more than one gene. The diagnosis of these syndromes is made by the characteristic clinical features specific to each disease, but the definitive diagnosis is achieved by laboratory investigations such as cytogenetic, biochemical and molecular methods. The importance of prenatal diagnosis and our experience are discussed in this article. Key-words: Ataxia telangiectasia, Bloom syndrome, Cockayne syndrome, Chromosome instability syndrome, Chromosome breakage, Chromosomal aberrations, DNA repair, Fanconi anemia, Nijmegan breakage syndrome, Trichothiodystrophy, Xeroderma pigmentosum. The definition of a chromosome instability syndrome is based on an increased frequency of spontaneous chromosome damage in the cells of the patients and strikingly high susceptibility to the chromosome damaging activity of specific mutagens. All these disorders show common clinical features of disturbances of growth and development, defects of the immune system and bone marrow function and a predisposition to develop malignant tumors. Most of these disorders are monogenic but different mutations in the genes can cause more than one disease phenotype. Recently cloning of a number of genes has been achieved. Cells are continually subjected to muta-genesis by various endogenous and exogenous agents due to which DNA gets damaged and changes in base pairs or breakages in DNA strands or DNA replication errors can occur. Damage to DNA consists of any deviation to its usual double helical structure. These changes can be single base changes or structural distortions. The mutagenic agents are ultraviolet radiations, ionizing radiations, alkylating agents and certain drugs. UV rays and alkylating agents cause base changes and cross linking of bases while ionizing rays produce free radicals causing mutations. Injury to DNA is minimized by the repair systems of the body which can recognize a range of distortions in the DNA and rectify the damage to DNA. There are a number of disorders charac-terized by an increased tendency of sponta-neous chromosomal breakages and are thus grouped into chromosomal breakage syn-dromes. In these syndromes the DNA repair mechanisms are defective resulting in the disease symptoms. The clinical features and diagnosis of each of these disorders is discussed separately. Table I lists the disorders described in the text. Table I:The Clinical Features and Gene Location of the Chromosomal Breakage Syndromes
This condition was first described by Fanconi in 1927 in 3 siblings. It is characterized by progressive pancytopenia, physical abnor-malities and a predisposition to malignancies. It involves a progressive failure in the bone marrow function. The cells are hypersensitive to DNA cross linking agents(1). Patients usually die at an early age. Phenotypically the patient shows pigmentation abnormalities, short stature and classic upper limb skeletal abnormalities in approximately 40% of cases. Four complementation groups have been identified (A,B,C,D), and different genes are responsible for these groups indicating heterogeneity in the disorder. The gene for group A is located on chromosome 20q and for group C on chromosome 9q 22.3. The sequence and molecular structure of Fanconi anemia complementation group C (FACC) is now known(2). Diagnosis: Fanconi anemia is clinically and genetically a heterogenous disease diagnosed on the basis of chromosomal instability to exposure of cultured cells with diepoxybutane (DEB)/mitomycin C or by carrying out sister chromatid exchanges (SCE) in subject samples. There is an increase in breakage frequency after treatment in such cases. Currently mutation analysis of the FA gene belonging to different complementation groups has been reported(3). Prenatal diagnosis can also be done by inducing fetal cells (chorionic villus samples, amnio-cytes, cord blood cells) by DEB/mitomycin C. It is important to perform studies of cellular sensitivity to DEB/mitomycin C in probands before performing prenatal diagnosis. Poor growth of amniocytes may be a useful indicator and used in conjunction with cytogenetic analysis while carrying out prenatal diagnosis. Now early prenatal diagnosis of FA is possible using DNA analysis(3,4).
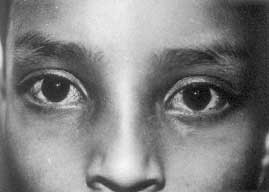
In 1941, Madame Louis-Bar reported this condition for the first time and Boder in 1975 described its pathology(5). It is a single gene multisystem disorder associated with prog-ressive neurodegeneration, characteristic telangiectases develop in the conjuctiva of the eyes with eventual development of lympho-reticular malignancy(6). It involves a complex spectrum of defects in the repair of lesions in DNA caused by ionizing radiations or chemical agents(7,8). In this disease following X-ray irradiation, alteration is seen in both semi-conservative replication and repair and there is a 6 to 10 fold increase in chromosomal break-ages. Serum alpha fetoprotein is increased, immunoglobulins IgA and IgG are deficient, motor delay is observed by 5 years of age, eye movement is abnormal by 3 years and 25% patients present with immunodeficiency symtpoms. Fig. 1 shows the clinical photograph of a patient with vascular telangiectasia. The AT locus was mapped to chromosome 11q 22-23 region(9) and the responsible ATM gene was subsequently identified(10,11). The mutated ATM gene is involved in DNA damage response at different cell cycle check points and also appears to have a wider role in signal transduction(12). Diagnosis: The diagnosis of ataxia telangiectasia can be made by cytogenetic analysis of the blood cells in patients and controls after exposing them to radiation and looking for higher frequency of chormosomal breakage and micronucleus assay. Prenatal diagnosis is done by analyzing chromosome breakages in the amniotic fluid cells in culture and observing their response to radiation. First trimester prenatal diagnosis of AT is also possible now biochemically by an assay of radioresistant DNA synthesis(3). Molecular studies have revealed 26 different muta-tions(14,15). The ability to recognize mutations across the entire coding sequence of the AT gene provides a practical advantage to AT families since a DNA based prenatal diagnosis is possible in families where the mutations are identified irrespective of the level of radio-sensitivity in these families.
It is a rare disorder in which the immuno-logical, cytogenetic and cell biological findings are similar to those found in ataxiatelangiectasia (AT). The disease is characterized by microcephaly, bird like face, short stature, chromosomal instability and immunodefi-ciency. It is also accompanied by recurrent infections and an increased predisposition to malignancies(16) and growth retardation(17). Mental development is normal in about 35% patients, borderline in 45% and moderate in about 20% patients. Immunodeficiency is severe, IgA deficiency is present as in AT but IgG deficiency is more common. Chromosomal instability and immunodeficiency predisposes the patient to malignancies. Matsura et al. mapped the gene mutated in NBS to human chromosome 8, using microcell mediated chromosome transfer. The gene has been localized on chromosome 8q 21-24. Two complementation groups V1 and V2 have been identified and this syndrome shares the cytogenetic and radiosensitivity characteristics of AT(18,19). Diagnosis: Cytogenetic analysis shows increased spontaneous and radiation-induced chromosomal breakage in about 10-35% of metaphases of cultured T-cells that frequently involves the immunoglobulin and T-cell receptor loci on chromosome 7 and 14(16,20). In a molecular genetic approach the features of radioresistant DNA synthesis (RDS) have been effectively used(13). For prenatal diagnosis two methods are available. The first method of assessment by radiation induced chromosome aberrations requires the analysis of large number of mitosis in chorionic villi (CV) and amniotic fluid (AF) cell cultures. The second method is radioresistant DNA synthesis. Both these methods are reliable and can be effectively used.
In 1954, Bloom described this condition for the first time(21). Clinically the patients show growth retardation and failure to thrive. They have a triangular face with telangiectatic rash on cheeks and areas exposed to sunlight. Females are fertile but men are infertile. Patients have high spontaneous frequencies of lymphatic and other malignancies, high baseline levels of SCE and high frequency of somatic cell mutation(22). Neither consistent hypersensitivity, other than that involving SCE’s(23) nor any defect in DNA repair has been demonstrated in Bloom’s syndrome(24). DNA ligase I is reported to have reduced activity and increased heat sensitivity in cells from patients with this syndrome(25). Bloom syndrome is a rare condition and the gene is localized on chromosome 15q21.3 and is a member of Rec Q helicase protein family which have the ability of unwinding DNA and RNA. The isolation of the gene for BS, known as the BLM, has also permitted the identi-fication of mutations within the gene. Diagnosis: Cytogenetically sister chromatid exchanges (SCE) or interchanges (between homologous chromosomes) take place with a greatly increased frequency in this disease. In addition an increased number of chromatid gaps, breaks and structural rearrangements of chromosomes are also seen in cultured cells. The cells are cultured in BrdU containing medium to enable differential staining and to study the exchanges between sister chromatids. Some molecular studies are also reported. Straughen et al. in 1998 have developed a rapid method for detecting restriction enzyme digestion of a PCR product containing the mutation(26). Here the blmAsh creates a restriction site within the amplified fragment allowing distinction of normal and mutant DNA.
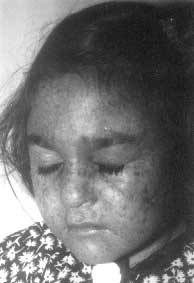
The predominant clinical feature of xeroderma pigmentosum is the increased sensitivity to ultraviolet rays present in sunlight. The skin is normal at birth but develops progressive atrophy, irregular pigmentation, telangiectasia and later keratoses, basal cell and squamous cell carcinomas. This syndrome is associated with a greater than 2000 fold increased frequency on sunlight induced skin cancers. The patients have a defect in the excision repair of UV radiation induced DNA damage. Progressive degenerative changes of exposed areas of the skin and eyes, often lead to neoplasia. Some patients also have progressive neurologic degeneration (18%). Age of onset of the disease symptoms is usually from 1-2 years but in 5% patients it is after 14 years of age. XP patients have defects in nucleotide excision repair (NER), the repair pathway that removes UV-induced damage(27). Fig. 2 shows the clinical picture of a patient with this disease. Genetic analysis of XP patients has revealed that there are 7 complementation groups (XPA to XPG), all showing deficiency in the excision repair and one variant type 1 identified for this disease. The 8 genes have been located and group A is lcoated on Chr 9, group B is located on Chr 2, group C on Chr 3, group D on Chr 19, group E on Chr 11, group F on Chr 16, group G on Chr 13 and XP variant type 1 on Chr 9(28). Diagnosis: The distinctive karyotype changes characteristic of other breakage syndromes are not seen in XP as usually normal karyotypes are found. Spontaneous and induced SCE can be visualized by culturing in BrdU containing medium staining with a photo-chemical reaction plus Giemsa(29). XP cells show a normal frequency of spontaneous SCE but a greater frequency after exposure of UV light or chemical carcinogens(30). UV induced rearrangements of DNA in form of SCE are elevated only in excision defective XP. Unscheduled DNA synthesis (UDS) is the classic method for diagnosis and requires 4 to 5 weeks before conclusion. The use of alkaline comet assay (single cell gel electrophoresis assay) is proposed as a simple repair test for earlier prenatal diagnosis(31). Recently, a Japanese group has carried out DNA based prenatal diagnosis for XP group A in a chorionic villus sample. It is a PCR based method followed by subsequent enzyme digestion to detect the three most frequent mutations of the XPA complementing genes (XPAc)(32).
Cockayne reported this disorder in 1946(33). It is multisystem sunsensitive genetic disorder in which a defect in DNA metabolism is seen in fibroblasts involving increased sensitivity to UV light and some chemicals(34). It is a premature aging syndrome. The repair to DNA damage is normal(35) but DNA replica-tion and RNA synthesis fail to recover from inhibition by UV damage. Microcephaly is accompanied by mental deficiency, unsteady gait and peripheral neuropathy. The CS cells lack a factor which is involved in targeting repair enzymes specifically towards DNA damage located in active DNA(36). The patients are specifically defective in the pre-ferential removal of damage from the trans-cribed strand of active genes, a process known as transcription-coupled repair, after UV irradiation. Two complementation groups (CSA and CSB) have been identified and about 80% of patients have been assigned to the CSB complementation group. The genes associated with both genetic complementation groups, CSA and CSB and a number of mutations in these groups have also been identified(33). Diagnosis: It has been observed that mutations in the ERCC6 gene are responsible for the Cockayne’s syndrome complementation group B, the most common form of the disease(37). A number of complementation group A mutations have also been identified, including a single base substitution that introduces a stop codon (322Tyr–> stop mutation in the C terminal region in both alleles of the CSA gene(38).
TTD is a rare autosomal recessive disorder and patients can be characterized by symptoms of sulphur deficient brittle hair, ichthyosis, physical and mental retardation, abnormal facies and in about 50% cases photosen-sitivity(39). There are no reports on association with skin cancers but patients have short life expectancy(40). The DNA repair characteristics of cells show a remarkable heterogeneity as some cells show normal repair, others show severe deficiency in nucleotide excision repair and some show a specific deficiency in the repair of UV induced photoproducts by nucleotide excision repair(41). Mutations in XP-D gene can also cause TTD. TTD patients fall into atleast 3 complementation groups, corresponding to TTD-A, XP-B, XP-D genes(42). Diagnosis: Diagnosis is made by studying the hair mounts (show the characteristic banding pattern with polarizing microscopy) and by hair aminoacid analysis (which demonstrates decreased high sulphur matrix proteins). Electron microscopic (EM) examination of keratinocytes show fibrillary bundles in the cytoplasm thinner and less electron dense than those of normal cells and large membrane bound vacuoles filled with granular filamentous material. Chromosomal DNA repair measured as the chromatid aberration frequency (CAF) after radiation shows that only one of TTD lines, TTD 1BR showed abnormally high CAF(43). In another report response to radiation in a patient was similar to XP-D cells and about 50% patients have photosensitivity and carry a defect in the nucleotide excision repair pathway similar XP-D patients(40,44). Complementation studies show that the defect is in the same genetic locus that underlies the cancer prone genetic disorder xeroderma pigmentosum (XP) group D(45,46). Molecular diagnosis of the disease has been reported by following RT-PCR and direct sequencing of the ERCC 2 locus.
Clinical observations suggest that XP, CS and TTD are a group of overlapping disease syndromes with dermatologic, neurological, neuropathological and other similarities including growth retardation and sexual immaturity. All XP and CS patients, and some TTD patients have sensitivity to UV radiations. Some patients have common symptoms of XP and CS while some others have TTD with symptoms like impaired intelligence and decreased fertility(39). The identification of mutations in the same gene with different symptoms of XP and CS/or TTD is reported(47,48). Mutations in XP-D gene can cause either XP, or XP and CS or TTD(42) whereas mutations in XP-G can cause XP or XP and CS(49).
There are few syndromes like Werner and Shwachman syndromes which do not fall under the classical definition of CBS but few studies have indicated that they also may be included in this category. Werner syndrome was described by Werner in 1904(50). The gene is called WRN and is mapped to chromosome 8p12. The mutations in this gene can lead to abnormalities of any function requiring un-winding of DNA. The disease is characterized by premature aging in young adults(51,52). Shwachman syndrome was described in 1963 by Shwachman(53). It is a rare multiorgan disease characterised by exocrine pancreatic insufficiency and bone marrow hypoplasia. Patients are predisposed to hematologic disorders(54,55) and immunodefi-ciency(56). They have short stature, failure to thrive and mental retardation. In these patients chromo-some studies show elevated frequencies of spontaneous chromosome aberrations com-pared with those in normal(57). Very few patients have been studied, therefore it is important to study chromosomal breakage in more patients with Shwachman and Werner syndrome to establish chromosome instability as a basic defect.
Now for many of these disorders prenatal diagnosis is possible. Fast growing chorionic villus cultures or amniotic fluid cell cultures are used in addition to a careful examination of cells of index patient. Prenatal diagnosis for AT, FA, NBS, BS and XP had been attempted successfully by the cytogenetic approach. Radioresistant DNA synthesis can also be used for NBS. Molecular methods have been used for diagnosing FA, BS, XP, CS and AT.
In our experience at the Genetic Unit, prenatal diagnosis was performed by cyto-genetic methods in two families of Fanconi’s anemia using two tests; DEB induced chromosomal breakage and micronuclei assay. In both cases normal fetuses were predicted. They were followed up at birth and were confirmed to be normal. Cytogenetic studies were also performed in ataxia telangiectasia homozygotes, obligate heterozygotes, controls and prenatal diagnosis was carried out in two fetuses at risk using amniotic fluid cultures. Both the cases of AT showed normal karyo-type(58). But significant differences were observed among the heterozygotes, homo-zygotes and controls. In one patient of xeroderma pigmentosum cytogenetic studies were carried out using BrdU staining and looking for sister chromatid exchanges. An increased frequency of SCE was observed showing that the patient was affected. Cytogenetic methods are conventionally used but are time consuming and labour intensive. There is always a risk for a culture failure or a contamination with bacteria or fungus growth. The need is to develop molecular techniques for quick and reliable diagnosis of these disorders.
|
![]()