|
|
Original Articles Indian Pediatrics 2000;37: 595-601 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Wilson's Disease - Early Onset and Lessons From a Pediatric Cohort in india |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Manuscript received:
March 30, 1999; Initial review completed: May 5, 1999;
Key words: Copper, Family screening, Wilson disease, Zinc. Wilson disease (WD) is an autosomal recessive condition characterized by inability of the liver to transport and store normally absorbed dietary copper resulting in abnormal deposition of copper in the basal ganglia, eyes, liver and other tissues. There is a great deal of variability in the clinical presentation; a typically hepatic form which presents in the first decade of life, neurologic symptoms generally due to extrapyramidal involvement and psychiatric symptoms which usually present later. In addition, hemolytic anemia, hypersplenism and renal tubular acidosis have all been described. This neccesitates exclusion of Wilson’s disease in a wide variety of symptoms, specially if they are familial. This retrospective analysis was carried out to determine the clinical and investigation profile, results of treatment and complications on follow up in patients with a diagnosis of Wilson’s disease in the Pediatric Department of our hospital.
A retrospective analysis of all children labelled as WD in case records of the Pediatric Neurology Clinic or the Pediatric Ward of the All India Institute of Medical Sciences, New Delhi, between 1986-1997 was carried out. Minimum criteria for the diagnosis of Wilson disease were the presence of a compatible clinical presentation, usually in the form of hepatic involvement or extrapyramidal neuro-logic involvement, Kayser Fleischer (KF) rings in the cornea confirmed by slit lamp examination and a 24 hour urine copper excre-tion greater than 100 micrograms or reduced serum ceruloplasmin level. Records were studied with particular reference to the age of onset of symptoms, time lag between onset of symptoms and diagnosis, and a family history suggestive of Wilson’s disease. Presenting symptoms were classified as either hepatic, neurologic, both, or other types of presenta-tions. Details of treatment received, and follow up parameters of patients were scrutinized to look for the effects of treatment, whether improved, deteriorated or unchanged. Wherever possible, patients were contacted to return for one more detailed follow up examination and laboratory workup which included a detailed general physical and neurologic examination, slit lamp examination of the cornea for KF rings. 24 hour urine copper estimation, liver function tests and screening for renal tubular acidosis. Complications, both of the disease and of treatment given, if any, were looked for. Screening of all siblings of index cases was carried out by an ophthalmic examination for KF rings and 24 hour urine copper excretion.
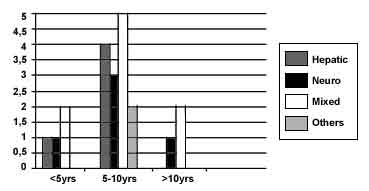
Demographics: Case records of 25 children, 14 males and 11 females, with the diagnosis of WD were identified in the years under review. Of these, 19 were index cases, and 6 others were asymptomatic (n = 4) or minimally symptomatic (n = 2) siblings of affected index cases who were diagnosed on screening of the family for Wilson’s disease giving us a total of 21 affected cases. The age of onset of initial symptomatology ranged from 3 years to 11 years with a mean of 7.2 years (Fig. 1). The mean time to presentation after the onset of symptoms was 2 years (range 2 months to 6 years). Family history: No history of consanguinity was obtained from any of the parents of affected children. Five index cases had family histories strongly suggestive of WD which were missed before contact in our hospital. These included seven sibling deaths from jaundice/cirrhosis and in one instance death from neurodegenerative disease suggestive of extrapyramidal involvement. Clinical presentation: The modes of presentation are listed in Table I. Eight children who later presented with extrapyramidal signs, gave a history of an acute self resolving hepatitis, clinically indistinguishable from viral hepatitis, in the one to two years preceding onset of neurologic illness. One child with hemolytic anemia presented with jaundice, hepatosplenomegaly, anemia (4.5 g/dl Hb), reticulocyte count of 4%, hemoglobinuria, serum bilirubin of 16 mg/dl with indirect bilirubin 8 mg/dl and raised liver enzymes (SGOT - 260 IU/1, SGPT - 360 IU/1, alkaline phosphatase 482 IU/1). Peripheral smear showed normocytic and normochromic anemia. Serum ceruloplasmin was 18 mg/dl. Liver biopsy showed chronic active hepatitis and increase in copper content. KF rings were negative. Twenty four hours urinary copper was 0.13 mg/24 hours. D-Penicillamine and zinc were started and after recovering from the attack of acute hemolysis, her liver enzymes continued to be high for 7 months after institution of treatment. An atypical presentation in another child was with a polyarthritis like manifestation consisting of pain, fever and swelling in multiple joints initially thought to be rheumatic fever, which was followed two years later by deteriorating school performance and altered handwriting which later progressed over the course of the next two years to frank extrapyramidal symptoms in the form of dystonia, dysarthria, tremors and a gait disorder. Family screening identified an additional six children with Wilson disease. Of these one had been noted to have symptoms of excessive drooling of saliva and tremors for the preceding three months and one had mild dysarthria; the remaining four were asymptomatic. Laboratory investigations obtained in these children included a 24 hour urine collection for copper estimation which ranged from 0.04 to 3.4 mg in 24 hours (normal <0.1 mg/24 h). Serum ceruloplasmin ranged from 12.3 to 19.0 mg/dl (normal >30 mg/dl). Liver function tests showed a mild elevation of serum transami-nases in one third of the children and prolonged prothrombin time in two thirds. Liver biopsies were performed in 3 patients only and revealed elevated stainable hepatic copper and evidence of chronic hepatic damage in 2. Neuroimaging in the form of CT or MRI was available in four children; one was normal, one showed a right sided hemiatrophy, one showed hypodensities in the white matter and bilateral thalami as well as cortical atrophy and one showed bilateral degeneration in the lentiform and caudate nucleus. Treatment in all cases was with D-penicillamine in a dose ranging from 8-30 mg/kg (average 15 mg/kg). It was given before food in 2 divided doses per day. Specific therapy was initiated after a time lag ranging from 2 months to six years after onset of initial symptomato-logy. In addition, all children received zinc (220 mg/day in 2 divided doses after meals) and supplementation with pyridoxine. Three children required phenothiazines intermittently for short periods of time to control exacerbation of extrapyramidal symptoms. Low copper diet and use of copper free utensils was also advised to all patients. Follow up (Table II) was available in 18 patients; follow up time ranged from 3 months to 10 years (3 mon-2 years in 6, 2-5 years in 7, 10 years in 5) following initial diagnosis. Most patients (n = 16) showed improvement on follow up with respect to extrapyramidal signs. Mild dysarthria persisted in 7 children. Hepatic complications included hepatic failure within 2-3 months after initiation of therapy with D- penicillamine in one child and upper GI bleeding in another. The patient with poly-arthritis at onset was rehospitalized on two occasions for recurrent fractures caused by severe osteopenia and renal rickets; investiga-tion at this time showed her to have renal tubular acidosis. She was put on Joule’s solution, vitamin D3 and calcium; however the patient continues to have severe osteopenia causing secondary degenerative arthropathy and loosening of teeth despite supplementation with vitamin D and calcium. The patient with hemolytic anemia at onset developed gallstones in the second year of follow up which remained asymptomatic with no further episodes of hemolysis. Side effects due to penicillamine were seen in only one child who developed a drug rash which responded to temporary discontinuation of the drug and coverage with steroids when it was reintroduced at a lower dose. Though KF rings became less intense and changed color to a brown discoloration they persisted on follow up and disappeared only in one. Two asymptomatic sibs on low dose penicillamine (8 mg/kg) and zinc developed KF rings in the fourth year of follow up.
Table I: Clinical Presentation
*
The patient with polyarthritis had associated neurologic symptoms. Table
II: Clinical Profile During Follow up (n=18) Duration
of follow up EPS -
extrapyramidal syndrome
Wilson’s disease is related to a derangement in copper metabolism, with high levels of liver copper and copper accumulation within the brain substance, especially within the central gray matter. It is transmitted as an autosomal recessive trait, with an incidence of 1 : 40000 to 1: 100,000(1) in homozygous form. Copper first accumulates in the liver; hence the usual presentation during childhood is that of hepatic disease. After the liver storage capacity for copper gets saturated, copper gets redistributed with accumulation in the nervous system, cornea, kidneys and other organs(2). Most patients present in the second decade of life with a primary hepatic presentation with the remainder of patients presenting during the third and fourth decade with a primarily neurologic or psychiatric presentation(3). Of note, in our series, the age of onset of symptoms (Fig. 1) for both the hepatic and neurologic groups was earlier than has been reported in other western series(4-7). The apparently early age of onset in our patient series could be partly attributed to the fact that ours is a pediatric unit and therefore skewed towards seeing younger patients. However, in other series of Wilson’s Disease from India(8-10), the age of onset was also lower. Thus, it is possible that the disease manifests at a younger age in Indian children. The average intake of copper in India ranges from 5.7-7.1 mg/day and is higher than the reported 0.34-1.1 mg/day in Western countries(11). The practice of cooking food in copper/copper alloy pots might be contributory(9-10). An analogous situation was reported by one of the authors for Indian Childhood Cirrhosis, a condition with heavy hepatic copper deposits(12). A history of a self resolving hepatitis was obtained from one third of our patients. In developing countries where viral hepatitis is endemic, it can often be difficult to distinguish viral from other metabolic causes of hepatitis. Hence, the index of suspicion needs to be high and the need to screen for Wilson’s disease should be considered in any case of atypical hepatitis. The need for family screening cannot be over emphasized. Almost one fourth of the patients in the present series were identified by family screening of index patients. A family history of jaundice/cirrhosis in a child with unexplained jaundice or neurological symptoms or coexistence of either in different sibs should make one strongly consider the diagnosis of WD. A slit lamp examination for the presence of KF rings appears a cost effective method to screen for Wilson’s disease. However, it should be kept in mind that false negative results have been described when using KF rings as a diagnostic index(13,14) and thus in any patient in whom clinical suspicion for Wilson disease is high, a 24 hour urine copper estimation should be done. A history of insidious alteration in behavior and school performance, preceding the onset of frank extrapyramidal symptoms was obtained in almost one fourth of our patients. Neuropsychiatric manifestations are well known to occur in Wilson disease(15) and in some instances psychiatric symptoms can be the first manifestation of disease and can obscure the diagnosis(16). Alterations in speech seem to be a sensitive indicator of extra-pyramidal symptomatology in Wilson disease as these were present in all patients with neurologic disease and was often the first symptom noted by parents. In three of our patients, mild dysarthria was the only neuro-logic abnormality noted on examination. Again on follow up examination, residual dysarthria was often the most persisent finding found after resolution of other more florid features such as tremor, dystonia and gait abnormalities. In a series of 136 patients with neurologic Wilsons disease, dysarthria and difficulty with hand movements were described as the earliest symptoms(7). Other neurologic findings in our patients with Wilson disease are similar to those described by other investigators(5,7) and essentially consisted of a wide variety of extrapyramidal symptomatology in the form of dystonia, tremors, hemiballismic movements, rigidity and choreiform movements. Side effects from penicillamine were less than has been reported in an other series(4). All our patients with Wilson’s disease, those with solely hepatic as well as those with neurologic disease except the patient with hemolytic anemia had KF rings on initial examination which persisted on follow up ophthalmologic examination in all. However, other investi-gators have reported that treatment with copper chelators results in gradual resolution of the rings over 3 to 5 years(17). It has been suggested that zinc be used for maintenance therapy and for treatment of asymptomatic sibs(18); however, 2 of our asymptomatic patients developed KF rings while on low dose penicillamine and zinc, suggesting that zinc alone may be inadequate as maintenance prophylactic therapy. The persistence of KF rings and dysarthria despite conventional doses of penicillamine and zinc might also imply the need for higher doses in our population in view of the dietary copper overload. In summary, Wilson’s disease should be strongly considered as a diagnostic possibility in any child with extrapyramidal neurologic symptoms. Alteration in speech, deterioration in school performance and change in behavior may often be the first symptoms noted. A previous history of jaundice, family history of jaundice or cirrhosis in siblings, increases the index of suspicion for WD exponentially. Family screening of index families with slit lamp examination, 24 hour urine copper excre-tion and ceruloplasmin levels cannot be overstressed.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![]()