Autoimmune
encephalitis (AIE) is being increasingly recognized as a
significant as well as frequent cause of encephalopathy in
the pediatric age group. Despite a plethora of antibodies
being described against the central nervous system, a
significant proportion of childhood auto-immune encephalitis
do not exhibit detectable known antibodies, spawning a
diagnostic challenge [1]. These children may have as yet
unidentified antibodies or other immune mechanisms. AIE
incorporates proven syndromes based on clinical
phenomenology and based on autoantibody associations. Of
these, syndromes with antibodies to cell surface antigens
have evidence to suggest pathogenicity. Rarely, antibodies
to intracellular antigens can be a biomarker but their role
is unproven.
The most common antibody associated
with AIE in children is anti-NMDA receptor (NMDAR) antibody.
Unlike adult AIE, association with cancer is less frequent
in children [2]. Early diagnosis and treatment leads to
better neurocognitive outcomes. Pediatricians and
intensivists need to be aware of this entity so that they
can ensure timely and appropriate diagnosis and treatment.
This review will provide readers with an updated account of
clinical presentation, diagnosis and treatment options in
autoimmune encephalitis in children, with discussion of
future priorities and challenges.
METHODS
A MEDLINE search
strategy using the following terms (1998-2019) was adopted
for this review. Limits of ‘Human’ and ‘English’ were
applied. Search terms included: “autoimmune encephalitis”,
“autoimmune encephalitis AND epidemiology”,
“pathophysiology”, “diagnosis” and “treatment” for studies
in children. Review articles, practice parameters,
guidelines, systematic reviews, meta-analyses, randomized
controlled trials, cohort studies, case series and case
reports were included.
EPIDEMIOLOGY
Data on the epidemiology of pediatric AIE is limited. A
retrospective study of anti-NMDAR encephalitis conducted
over seven years in Hong Kong estimated an incidence of 2.2/
million children per year [3]. This disorder likely accounts
for a large number of cases of encephalitis in children.
Anti-NMDAR encephalitis may also contribute to recurrence of
encephalitis following herpes simplex virus encephalitis in
both children and adults [4]. Other non-herpes viruses may
also act as triggers for anti-NMDAR encephalitis [5].
Anti-NMDAR encephalitis accounts for 4% of all encephalitis
and is the most common cause of seropositive AIE in
children. Almost 40% of all reported cases are below 18
years of age [6]. Steroid responsive encephalopathy
associated with autoimmune thyroiditis (SREAT) or
Hashimoto’s encephalopathy has a prevalence of 2/100,000 in
adults but its frequency in children is much less [7].
Anti-thyroid antibodies may be detected in up to 10% of
normal children, entailing caution while interpreting these
in the presence of neurological impairment in children [8].
PATHOGENESIS
Autoimmune encephalitis can be categorized as per antigen
location into two groups. In one group, antibodies target
intracellular antigens, and in the second, antibodies target
cell surface antigens. This categorization has clinical
relevance as well. Intracellular antigen-based diseases are
usually paraneoplastic and are mediated by cytotoxic T-cells
[9]. These syndromes respond poorly to immunomodulatory
therapy with poorer outcomes [10]. Cell surface
antigen-based diseases have a lower association with
malignancy and are mediated by the humoral immune system
[11]. These have a better response to immunotherapy and a
more favourable outcome [12]. AIE may also be paraneoplastic
or non-paraneoplastic, based on the presence or absence of
an underlying neoplasm, respectively - although, this is
less relevant in pediatric AIE.
Paraneoplastic syndromes: These
result when tumor antigens are shared by neuronal cell
antigens, leading to antibody-mediated immunological
destruction of neural tissue [13,14].
Infections:
Another mechanism is post-viral autoimmune encephalitis.
This was first highlighted in a study that reported the
development of anti-NMDAR antibodies in 30% of patients with
HSV encephalitis based on CSF PCR studies [15]. It is now
known that relapsing symptoms following HSV encephalitis
that lack viral antigen positivity may be attributable to
anti-NMDAR antibodies in 20% of the cases, with a higher
frequency in children [16]. These relapses improve
dramatically with immune therapy. A putative mechanism
involves the release of brain-specific neo-antigens caused
by viral toxicity that trigger development of pathogenic
anti-bodies. Another mechanism may be the non-specific
stimulation of a range of antibodies following viral
inflammation. In children, these relapses frequently take
the form of choreo-athetosis and diminished conscious-ness.
Even the viral phase of herpes virus encephalitis may have
immunological basis, supported by the occurrence of less
severe disease in immunocompro-mised individuals [17], as
well as the beneficial role of steroid therapy in this
condition [18]. Although less frequently documented, other
viral infections such as varicella zoster, Epstein Barr
virus (EBV), Human herpes virus-6 (HHV-6), Cytomegalo-virus
(CMV), adenovirus, rickettsial infection as well as HIV are
also known to predate AIE [19]. Non-NMDAR antibodies have
also been reported after viral encephalitis, including
anti-D2 receptor, anti-GABA-A/B, anti-AMPAR antibodies [20].
Post-vaccinal:
Several cases of anti-NMDAR encephalitis have been reported
following vaccination with influenza (H1N1), diphtheria,
tetanus, pertussis, polio and Japanese B encephalitis
vaccination [21].
CLINICAL FEATURES
Pediatric
autoimmune encephalitis clinically manifests as various
clinical syndromes dictated by the type of antibody. Both
paraneoplastic and non-paraneoplastic syndromes are
associated with the following broad type of antibodies: (i)
antibodies directed against cell-surface antigens, (ii)
antibodies directed against intracellular antigens, and (iii)
antibodies directed towards synaptic antigens present on the
extracellular surface. The clinical syndromes are summarized
in Web Table I.
Anti-NMDAR
Encephalitis
Anti-NMDAR
encephalitis accounts for 4% of all encephalitis and is the
most common cause of seropositive AIE in children. This
entity was first described in 2007 as a paraneoplastic
syndrome in adult females in association with ovarian
teratomas [22]. Since then, it has been described in men,
women and children of all age groups, with and without
teratomas. Almost 40% of all reported cases are below 18
years of age [6]. Pathogenic IgG1 antibodies bind to the
GluN1 subunits of the N-Methyl-D-aspartic acid receptor
leading to their internalization. Clinical features include
a prodrome in 50% of cases lasting weeks to months
comprising fever, malaise, headache, gastrointestinal or
respiratory comp-laints followed by neurological (abnormal
behavior, cognitive deterioration, short-term memory loss,
seizures, movement disorders, central hypoventilation
syndrome), psychiatric (delusions, hallucinations,
catatonia) and autonomic dysfunction [23,24]. Younger
patients tend to present with seizures and movement
disorders compared to adults who present with psychiatric
abnormalities [22]. Children with anti-NMDAR encephalitis
have multiple symptoms, and monosymptomatic cases are
present in only 1% of patients which is why, anti-NMDAR
encephalitis is unlikely to be a cause of isolated psychosis
and is usually accompanied by seizures [23]. Seizures, seen
in up to 80% of patients, may be focal or generalized,
including status epilepticus, and may occur in any stage of
the disease [23]. In a study from New Delhi of 15 patients
with AIE (age range 2-64 years), seizures were reported in
100% patients [24]. Movement disorders include orofacial
dyskinesias, chorea-athetosis, ballismus, rigidity,
opisthotonus and tremors [25]. Advanced disease is
characterized by stupor, coma, periods of agitation
alternating with catatonia as well as autonomic dysfunction.
In younger children, behavioral changes may be difficult to
discern as they present with temper tantrums, irritability
and hyperactivity as opposed to frank psychosis. Unlike
adults, the first symptoms are non-psychiatric, ranging from
dystonia and seizures to mutism. In a study from Chandigarh
that studied patients below 12 years of age with anti-NMDAR
encephalitis, the presence of extreme irritability, insomnia
and mutism were reported in all the children [26]. Three
clinical phenotypes have been described viz., the
classic form and the psychiatric form (associated with good
outcomes) and the catatonia-predominant form (associated
with poor outcome) [27].
Atypical forms have also been
described with children presenting with dominant autistic
regression [28], catatonia and neuroleptic malignant
syndrome [29] and gait disorder [30]. The presentation of
pediatric anti-NMDAR encephalitis differs from adult AIE in
several respects and these are summarized in Table
I.
Table I Clinical Features of Anti-NMDAR Encephalitis in Children and Adults
|
Clinical features |
Adults |
Children |
|
Initial feature |
Change in mood and behavior, psychosis |
Seizures, movement disorders, speech abnormalities, sleep problems |
|
Features at nadir of |
Seizures, impaired memory, movement |
Seizures, movement disorders, change of behavior |
|
disorders, impaired consciousness | |
|
Autonomic dysfunction |
Arrhythmia, central hypoventilation |
Tachycardia, hyperthermia, hypertension |
|
Association |
Tumors, post-infective |
Post-infective |
Overlapping Encephalitis
A recent study
showed that some patients with anti-NMDAR encephalitis had
an overlap in terms of clinical features or magnetic
resonance imaging (MRI) findings with neuromyelitis optica
(NMO) [31]. Syndromes with dual-positive antibodies
have also started to be recognized, for e.g.
anti-NMDAR and anti-MOG or anti-AQP4 or anti-D2 receptor
positivity, anti-GAD and anti-GABA-A etc [32]. The
proportion of anti-GABA-B antibodies with overlap seem to be
more. Among 20 patients with anti-GABA-B receptor
encephalitis, seven showed overlap with other antibodies
[33]. Anti-NDMAR encephalitis may also overlap with
opsoclonus syndrome [34].
Seronegative Autoimmune Encephalitis
Only up to 44% of
patients with AIE have an antibody-positive status [1].
‘Seronegative but suspected autoimmune encephalitis’ has
received a consensus definition [1]. The definition includes
rapid progression of symptoms, along with exclusion of
well-defined AIE syndromes such as typical limbic
encephalitis, absence of serum and CSF antibodies along with
two of: MRI abnormalities suggestive of autoimmune
encephalitis, CSF pleocytosis, CSF-specific oligoclonal
bands or elevated CSF IgG index or brain biopsy showing
inflammatory infiltrates, along with exclusion of other
causes [35].
When to Suspect
Autoimmune Encephalitis?
The diagnosis of
AIE should be suspected in all children who develop a
polysymptomatic syndrome encom-passing encephalopathy,
seizures, movement disorders, psychiatric features, gait
disturbances and autonomic disturbances. The clinical
features suggestive of autoimmune encephalitis include:
•
Abrupt onset / rapid decline
•
Autonomic instability
•
Delirium slipping into catatonia and vice versa
•
Urinary/ faecal incontinence
•
Cognitive slowing
•
Gait and balance disorder
•
Relapse after treatment for viral encephalitis
•
Seizures that may be in the form of status epilepticus or
multifocal drug resistant epilepsy or seizure clusters
•
Involvement of multiple domains eg. Cognition and
extrapyramidal system etc.
• CSF
may also reveal features of inflammation in the absence of
infection.
Features that point away from the diagnosis of AIE include:
• A
very chronic or indolent course
•
Plateauing of symptoms
• No
impairment in activities of daily living
•
Cognition remaining intact
•
Purely psychiatric symptoms
Table
II
depicts some differentiating features between autoimmune and
infective (viral) encephalitis.
DIAGNOSIS
The diagnosis of AIE is based on the presence of an
appropriate clinical syndrome supported by various ancillary
investigations. All other possible etiologies should be
ruled out along with confirmatory antibody testing. The
common differentials include CNS infections, toxins, CNS
vasculitis, inborn errors of metabolism, neoplasms and a
primary psychiatric disorder. The features supportive of an
autoimmune etiology include evidence of CNS inflammation
(CSF pleocytosis, elevated IgG index or oligoclonal bands,
elevated CSF neopterin), MRI abnormalities and a response to
immunosuppressive treatment. Criteria for the diagnosis of
anti-NMDAR encephalitis have been proposed by Graus, et
al. [35]. As per this criteria, probable anti-NMDAR
encephalitis can be made if all three of the following
criteria have been met: (i) Rapid onset (less than
three months) of at least four symptoms among
psychiatric/behavioral dysfunction, speech abnormalities,
seizures, movement disorders, decreased consciousness or
autonomic dysfunction; (ii) Abnormal EEG/CSF; and (iii)
Exclusion of other causes. A study evaluating the
reliability of these criteria found them to be 90% sensitive
and 96% specific for the diagnosis of anti-NMDAR
encephalitis in children [36]. Diagnostic evaluation
includes the following:
Magnetic Resonance Imaging of Brain
Classic neuroimaging abnormalities in AIE include unilateral
or bilateral T2/ FLAIR signal hyperintensities involving the
mesial temporal lobe. The large majority of patients (66%)
with anti-NMDAR encephalitis do not exhibit neuroimaging
abnormalities [37]. Abnormalities in the form of signal
hyperintensities may be seen throughout the brain.
Transitory cortical enhancement in the absence of restricted
diffusion or hemorrhage may also be seen [37]. In patients
with normal MRI and typical clinical and EEG picture,
positron emission tomography may be useful to highlight
involvement of the mesial temporal lobes [38]. In contrast
to NMDAR encephalitis, the large majority of patients with
limbic encephalitis such as anti-Lgi1 antibodies exhibit
mesial temporal hyperintensities and may go on to develop
mesial temporal sclerosis on follow-up imaging [39]. The
presence of restricted diffusion and contrast enhancement
correlated with the development of mesial temporal sclerosis
[39]. MRI is mostly abnormal in anti-GABA-A receptor and
anti-D2 receptor encephalitis. Most patients with
anti-GABA-A receptor encephalitis show MRI abnormality in
the form of extensive, multifocal or diffuse cortical and
subcortical T2/FLAIR signal alterations. Rapid progression
from frontal and temporal T2/FLAIR abnormalities to atrophy
and extensive bilateral lesions has been reported in some
patients. Majority of patients with anti-D2 receptor
encephalitis exhibit bilateral basal ganglia T2/FLAIR signal
abnormalities.
Electroencephalography
EEG may show focal or diffuse slowing as well
as epileptiform discharges. 30% of anti-NMDAR
encephalitis patients may exhibit a typical pattern called
‘extreme delta brush’ [40].
Antibody Testing
Confirmation of the pathogenic antibody forms the basis for
diagnosis of autoimmune encephalitis. Those testing positive
are deemed ‘definite’ cases, while those who do not are
labelled ‘suspected’. These antibodies bind to
conformational extracellular epitopes of proteins on the
cell surface like receptors, synaptic proteins or ion
channels. Their shape and conformation determine antibody
binding. Therefore, cell-based assays with live or fixed
eukaryotic cells should be used. The importance of the same
was highlighted in the false positivity associated with
voltage gated potassium channel (VGKC) complex
radioimmunoassay because it not only precipitates the target
antigens: leucine rich glioma inactivated (LGI1) and contact
in associated protein 2 (CASPR2) but also other
intracellular antigens [41]. Serum testing for these
antibodies is non-inferior to CSF testing, except in the
case of anti-NMDAR encephalitis, where CSF testing is more
sensitive [42] with CSF sensitivity being 100% (versus 85.6%
in serum). In addition, commercial anti-NMDAR testing should
be done using assays that test IgG antibodies to the
extracellular domain of the NR1 subunit of the receptor.
Antibodies such as serum IgA or other antibody types other
than IgG, or antibodies to the NR2 subunit, do not
necessitate treatment as these are not clinically relevant.
Testing both serum and CSF should be done whenever possible.
The utility of follow up evaluation of these antibodies has
not yet been ascertained and is therefore not indicated as
of now. If diagnosis is delayed or patients have received
treatment with plasma exchange or IV immunoglobulin,
antibodies might be detected only in CSF. Patients with a
protracted clinical course or persistent symptoms might be
sero-negative and have persistently raised CSF titres until
symptoms improve [43]. Less frequently, long-term follow-up
reveals patients who, after recovery, still have high serum
titers and absent or barely detectable titers in the CSF.
Findings are consistent with a disease in which the immune
response is initially triggered systemically by a tumor or
other unknown causes and is reactivated and expanded in the
CNS.
TREATMENT
Basic tenets that guide the treatment of autoimmune
encephalitis are that patients treated with immunotherapy
fare better than those not given immunotherapy. Earlier
initiation of immune therapy is associated with better
prognosis. Lastly, if the patient does not respond to first
line therapy, or if the disease is severe or relapsing,
treat-ment with a second-line agent improves prognosis [44].
The primary immunomodulation options
include steroids, intravenous immunoglobulins or plasma
exchange. This may be followed by maintenance therapy in the
form of oral steroid taper, monthly pulse steroids or pulse
IVIG therapy. Azathioprine and mycophenolate mofetil are
often used in maintenance therapy as steroid-sparing agents.
Usual duration of maintenance therapy ranges from 6 to 12
months but is individualized. Second line therapy in case of
non-response to first line agents includes rituximab.
Cyclophosphamide is another second line agent. Third line
agents include bortezomib and tocilizumab. Another important
tenet is to screen for tumours, especially in adolescent
females, due to the association with ovarian teratomas.
Additionally, clinicians must consider Subacute sclerosing
panencephalitis (SSPE) in the differential as it is a close
mimic of AIE, presenting as cognitive decline, seizures,
myoclonic jerks, ataxia and extrapyramidal disorders.
First-line Therapy
Corticosteroids
form the cornerstone of treatment. They have good
penetration across the blood brain barrier and have a broad
spectrum of anti-inflammatory activity. They are usually
given as a pulse therapy with methylprednisolone (30 mg/kg/d
for 3-5 days, maximum 1g/d), followed by sustained oral
steroids according to bodyweight (prednisolone 1-2 mg
kg/day) followed by slow taper over 6-12 months (in severe
syndromes like anti-NMDAR encephalitis), determined by
case-based scenario. Intravenous immunoglobulin (IVIG) (2
g/kg given over 5 days) or plasma exchange (PLEX) (5 to 7
exchanges of 50 mL/kg every alternate day) are commonly used
as alternatives and occasionally, concomitantly. Evidence,
although scarce, has found early PLEX along with
corticosteroids to have better outcomes than either alone
[45]. No evidence exists regarding the superiority of PLEX
versus IVIG. However, considering that patients with
autoimmune encephalitis are commonly agitated, IVIG might be
easier to administer. In a study from Bangalore, 13 children
with anti-NMDAR encephalitis were followed up for a mean
duration of 10.3 (6.7) months [46]. All patients were
administered intravenous methylprednisolone followed by
monthly pulses of methyl prednisolone. IVIG and PLEX were
administered during the acute phase for inadequate response
to methyl prednisolone. The study concluded that Anti-NMDAR
encephalitis required prolonged immunomodulatory therapy and
methylprednisolone was effective for this purpose [46].
If AIE is suspected, empirical therapy has to be initiated
immediately. Waiting for the results of antibody tests is
not an essential pre-requisite. If resources are a
constraint, CSF is the preferred sample for antibody testing
because it is more sensitive than serum, especially for
anti-NMDAR encephalitis [42]. If the patient is unable to
afford antibody testing altogether, empirical therapy should
be initiated after reasonably excluding alternate causes.
Fig. 1 depicts a diagnostic and therapeutic
algorithm in children with suspected AIE.
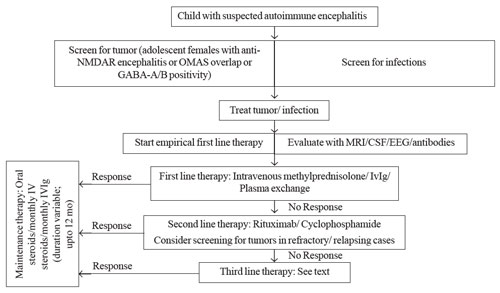 |
| Fig. 1
Suggested management algorithm for a child with
suspected autoimmune encephalitis. |
Second-line Therapy
A significant proportion of patients respond to first line
therapy, showing benefit of treatment within the first 1-2
weeks of treatment initiation. Non-responders are treated
with 2nd line
agents viz, rituximab or Cyclophosphamide. Rituximab
is a chimeric monoclonal antibody against CD20 resulting in
B-cell depletion, which leads to reduced pro-inflammatory
CD4+ and CD8+ T cell responses [47].
B-cell measurement should be done 2-4 weeks after dosing to
check for B-cell depletion (some children may be resistant
which entails a substitute treatment) and 3-6 monthly
thereafter to look for B-cell repopulation, that may assist
redosing, if clinical symptoms persist or a relapse is
suspected [48]. Although well tolerated, infusion reactions
occur in approximately 12% individuals, and serious adverse
events are rare. Dale, et al. reported serious
adverse events in 4 children out of the 144 treated with it,
including two deaths [49].
Cyclophosphamide is the other
alternative and has broad cellular immune suppression
effects. Monthly intravenous infusions of 500-1000 mg/m2 body
surface area for 6-9 months is the usual course of
treatment.
The risks of infertility and secondary malignancies
are the major limitations to its use. However, these are
dependent on the cumulative dose received and doses <7.5 g/m2 are
justified in sick patients. In the case
series of Dale et al [49], 58 of the 144 patients
received concomitant rituximab and cyclophosphamide
without any increase in the adverse effect profile. This
provides re-assurance for using both together, if the need
arises.
Third-line Agents
Third line agents are needed when both 1st and
2nd line
agents fail. Literature regarding their use is limited to
general recommendations. Behrendt, et al. [50] showed
benefit of Bortezomib (protease inhibitor which inhibits the
pro-inflammatory signalling cascade) in two adults with
severe refractory anti-NMDAR encephalitis. Tocilizumab, an
anti-IL6, has also been tried [51] based on the observation
of elevated levels of IL6 in the CSF of patients with
anti-NMDAR and anti-MOG associated disease [52]. Tatencloux,
et al. have used intrathecal steroids and
methotrexate in pediatric patients with refractory
anti-NMDAR encephalitis.
Maintenance Therapy
Mycophenolate mofetil (MMF), methotrexate and azathioprine
have been used as steroid-sparing agents in paediatric
anti-NMDAR encephalitis. In a systematic review of
retrospective cohort data, MMF/ methotrexate/ azathioprine
used individually or in varying combinations were associated
with a reduced risk of relapse if started after the first
event rather than after subsequent ones, and were reasonably
safe [54].
Other Measures
Symptomatic therapy: Symptomatic
management should be given along with immunosuppressive
treatment. Sedating agents are used to induce and maintain
sleep, relieve agitation and emotional imbalance.
Benzodiazepines, anti-epileptics, clonidine and chloral
hydrate are commonly used for this purpose. Neuroleptics are
best avoided due to the high incidence of adverse effects
like rigidity and neuroleptic malignant syndrome.
Management of relapses: Relapses
tend to be uncommon in AIE. However, when they do occur,
they are managed with repeat dosing of the first line
agents. In these cases, there is concern of ongoing
inflammatory activity, and hence, chronic immunosuppressive
therapy such as azathioprine, mycophenolate or repeated dose
of rituximab may be considered.
PROGNOSIS
Most patients with
anti-NMDAR encephalitis respond to immune therapy. A study
with a median follow up of 24 months showed that 94%
patients responded within four weeks to first line
immunotherapy/ tumour removal [23]. Of the patients who
failed first line therapy, 57% underwent second line therapy
and had better outcomes. At 24 months follow up, 81%
patients had a good outcome, with mortality in 6%. Outcomes
continued to improve up to 18 months following treatment.
Predictors of good outcome included early treatment and lack
of intensive care unit admission.
Relapses in AIE tend to be uncommon
and the approximate percentage varies according to the
subtype being dealt with. Approximately 12% of patients with
anti-NMDAR encephalitis were found to relapse in initial
descriptions [42]. However, this has reduced, probably due
to the use of second line therapies and chronic
immunosuppression, which lead to the alteration in the
natural history of disease. The patients that do relapse
tend to be mono-symptomatic, presenting with seizures or
movement disorders commonly, unlike the initial presentation
which almost always tends to be polysymptomatic. Chronic
immunosuppression with mycophenolate, azathioprine or
re-dosing with rituximab is done in this scenario.
FUTURE DIRECTIONS
Pediatric
autoimmune encephalitis is a challenging condition to
diagnose and treat and these are suffers from several
lacunae in evidence. More literature is required on the
diagnosis of suspected autoimmune encephalitis in children
with seronegativity as well as on overlap syndromes.
Duration of optimal therapy in children is also not clear.
Another challenging aspect of therapy that demands research
is the management of refractory autoimmune encephalitis.
However, it is heartening that with the current status of
knowledge, appropriate and timely management can ensure
satisfactory outcomes in the majority.
Contributors:
All the authors were involved in the concept and design of
the article. DG: performed the literature review and wrote
the first draft which was revised by SS and SSM. All authors
approve the final draft of the manuscript submitted for
publication.
Funding:
None; Competing interest: None stated.
|
Key Messages |
•
Pediatric autoimmune encephalitis forms a group of
acquired disorders with antibodies targeting
cell-surface antigens or intracellular antigens that
are treatable.
• Pediatric disease manifests
differently from adults, with less frequent
association with neoplasms and predominance of
movement disorders, behavioral abnormalities and
seizures.
• Anti-NMDAR encephalitis is the
most common pediatric autoimmune encephalitis. It
exhibits typical clinical features (limbic
encephalitis) as well as imaging abnormalities
(mesial temporal signal change) although these may
be seen in only 30-40% of patients. Hence, clinical
recognition is the key. It responds well to early
therapy.
• Treatment involves
immunomodulation which should be initiated
empirically as soon as the diagnosis of autoimmune
encephalitis is suspected, even prior to the
availability of antibody test results.
|
REFERENCES
1. Hacohen Y, Wright S,
Waters P, Agrawal S, Carr L, Cross H, et al.
Paediatric autoimmune encephalopathies: clinical features,
laboratory investigations and outcomes in patients with or
without antibodies to known central nervous system
autoantigens. J Neurol Neurosurg Psychiatry. 2013;
84:748-55.
2. Armangue T,
Petit-Pedrol M, Dalmau J. Autoimmune encephalitis in
children. J Child Neurol. 2012; 27:1460-1469.
3. Ho AC, Chan SH, Chan
E, Wong SS, Fung ST, Cherk SW, et al.
Anti-N-methyl-d-aspartate receptor encephalitis in children:
Incidence and experience in Hong Kong. Brain Dev. 2018;
6:473-9.
4. Peery HE, Day GS,
Doja A, Xia C, Fritzler MJ, Foster WG, et al.
Anti-NMDA receptor encephalitis in children: the disorder,
its diagnosis, and treatment. Hand Clin Neurol.
2013;119:1229-33.
4. Glaser CA, Honarmand
S, Anderson LJ, Schnurr DP, Forghani B, Cossen CK, et al.
Beyond viruses: Clinical profiles and etiologies associated
with encephalitis. Clin Infect Dis. 2006;43:1565-77.
6. Florance NR, Davis
RL, Lam C, Szperka C, Zhou L, Ahmad S, et al.
Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in
children and adolescents. Ann Neurol. 2009;66:11-18.
7. Ferracci F, Bertiato
G, Moretto G. Hashimoto’s encephalopathy: Epidemiologic data
and pathogenetic considerations. J Neurol Sci.
2004;217:165-8.
8. Zois C, Stavrou I,
Kalogera C, Svarna E, Dimoliatis I, Seferiadis K, et al.
High prevalence of auto-immune thyroiditis in school
children after elimination of iodine deficiency in
northwestern
Greece. Thyroid. 2003;13: 485-9.
9. Bien CG, Vincent A,
Barnett MH, Becker AJ, Blumcke I, Graus F, et al.
Immunopathology of autoantibody-associated encephalitides:
Clues for pathogenesis. Brain. 2012;135:1622-38.
10. Dalmau J, Bataller
L. Clinical and immunological diversity of limbic
encephalitis: A model for paraneoplastic neurologic
disorders. Hematol Oncol Clin North Am. 2006;20:1319-35.
11. Graus F, Saiz A,
Dalmau J. Antibodies and neuronal autoimmune disorders of
the CNS. J Neurol. 2010;257: 509-17.
12. Lancaster E,
Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies
to synaptic and neuronal cell surface proteins. Neurology.
2011;77:179-89.
13. Gultekin SH,
Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J, et
al.
Paraneoplastic limbic encephalitis: neurological
symptoms, immunological findings and tumour association in
50 patients. Brain. 2000;123:
1481-94.
14. Dalmau J, Lancaster
E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R.
Clinical experience and laboratory investigations in
patients with anti-NMDAR encephalitis. Lancet Neurol.
2011;10:63-74.
15. Pruss H, Finke C,
Holtje M, Hofmann J, Klingbeil C, Probst C, et al.
N-methyl-D-aspartate receptor antibodies in herpes simplex
encephalitis. Ann Neurol. 2012;72:
902-11.
16. Pruss H. Postviral
autoimmune encephalitis: manifesta-tions in children and
adults. Curr Opin Neurol. 2017; 30; 327-30.
17. Sellner J, Dvorak
F, Zhou Y, Haas J, Kehm R, Wildemann B, et al. Acute
and long-term alteration of chemokine mRNA expression after
antiviral and anti-inflammatory treatment in herpes simplex
virus encephalitis. Neurosci Lett. 2005; 374:197-202.
18. Kamei S, Sekizawa
T, Shiota H, Mizutani T, Itoyama Y, Takasu T, et al.
Evaluation of combination therapy using aciclovir and
corticosteroid in adult patients with herpes simplex virus
encephalitis. J Neurol Neurosurg Psychiatry. 2005;76:1544-9.
19. Mohammad SS,
Sinclair K, Pillai S, Merheb V, Aumann TD, Gill D, et al.
Herpes simplex encephalitis relapse with chorea is
associated with autoantibodies to N-Methyl-D-aspartate
receptor or dopamine-2 receptor. Mov Disord. 2014;29:117-22.
20. Armangue T, Moris
G, Cantarin-Extremera V, Conde CE, Rostasy K, Erro ME, et
al. Spanish Prospective Multicentric Study of
Autoimmunity in Herpes Simplex Encephalitis. Autoimmune
postherpes simplex encephalitis of adults and teenagers.
Neurology. 2015; 85:1736-43.
21. Wang H. Anti-NMDA
receptor encephalitis and vaccination. Int J Mol Sci.
2017;18:193.
22. Dalmau J, Tuzun E,
Wu HY, Masjuan J, Rossi JE, Voloschin A, et al.
Paraneoplastic anti-N-methyl-D-aspartate receptor
encephalitis associated with ovarian teratoma. Ann Neurol.
2007;61:25-36.
23. Titulaer MJ,
McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T,
et al. Treatment and prognostic factors for long-term
outcome in patients with anti-NMDA receptor encephalitis: an
observational cohort study. Lancet Neurol. 2013;12:157-65.
24. Pandit AK, Ihtisham
K, Garg A, Gulati S, Padma MV, Tripathi M, et al.
Autoimmune encephalitis: A potentially reversible cause of
status epilepticus, epilepsy and cognitive decline. Ann
Indian Acad Neurol 2013;16;
577-84.
25. Baizabal-Carvallo
JF, Stocco A, Muscal E, Jankovic J. The spectrum of movement
disorders in children with anti-NMDA receptor encephalitis.
Mov Disord. 2013;28:543-7.
26. Suthar R, Saini AG,
Sankhyan N, Sahu JK, Singhi P. Childhood anti-NMDA receptor
encephalitis. Indian J Pediatr. 2016;83:628-33.
27. Desena AD,
Greenberg BM, Graves D. Three phenotypes of
anti-n-methyl-d-aspartate receptor antibody encephalitis in
children: prevalence of symptoms and prognosis. Pediatr
Neurol. 2014;51:542-9.
28. Hacohen Y, Wright
S, Gadian J, Vincent A, Lim M, Wassmer E, et al.
N-methyl-d-aspartate (NMDA) receptor antibodies encephalitis
mimicking an autistic regression. Dev Med Child Neurol.
2016;58:1092-4.
29. Kiani R, Lawden M,
Eames P, Critchley P, Bhaumik S, Odedra S, et al.
Anti-NMDA-receptor encephalitis presenting with
catatonia and neuroleptic malignant syndrome in patients
with intellectual disability and autism. Br J Psych Bull.
2015;39:32-5.
30. Yeshokumar AK, Sun
LR, Klein JL, Baranano KW, Pardo CA. Gait disturbance as the
presenting symptom in Anti-NMDA receptor encephalitis.
Pediatrics. 2016;138: e20160901.
31. Titulaer MJ,
Hoftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T,
et al. Overlapping demyeli-nating syndromes and
anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol.
2014;75:411-28.
32. Gresa-Arribas N,
Arino H, Martinez-Hernandez E, Petit-Pedrol M, Sabater L,
Saiz A, et al. Antibodies to inhibitory synaptic
proteins in neurological syndromes associated with glutamic
acid decarboxylase autoimmunity. PLoS One. 2015;10:
e0121364.
33. Höftberger R,
Titulaer MJ, Sabater L, Dome B, Rózsás A, Hegedus B, et
al. Encephalitis and GABAB receptor antibodies: Novel
findings in a new case series of 20 patients. Neurology.
2013;81:1500-6.
34. Kurian M, Lalive
PH, Dalmau JO, Horvath J. Opsoclonus-myoclonus syndrome in
anti-N-methyl-D-aspartate receptor encephalitis. Arch
Neurol. 2010;67:118-21.
35. Graus F, Titulaer
MJ, Balu R, Susanne Benseler, Bien
CG, Cellucci T, et al. A clinical approach to
diagnosis
of autoimmune encephalitis. Lancet Neurol. 2016;15:
391-404.
36. Ho ACC, Mohammad
SS, Pillai SC, Tantsis E, Jones H, Ho R, et al. High
sensitivity and specificity in proposed clinical diagnostic
criteria for anti-N-methyl-d-aspartate receptor
encephalitis. Developmental Medicine and Child Neurology.
2016;59:1256-60.
37. Irani SR, Bera K,
Waters P, Zuliani L, Maxwell S, Zandi MS, et al.
N-methyl-D-aspartate antibody encephalitis: temporal
progression of clinical and paraclinical observations in a
predominantly non-paraneoplastic disorder of both sexes.
Brain. 2010;133:1655-67.
38. Baumgartner A,
Rauer S, Mader I, Meyer PT. Cerebral FDG-PET and MRI
findings in autoimmune limbic encephalitis: correlation with
autoantibody types. J Neurol. 2013; 260:2744-53.
39. Kotsenas AL, Watson
RE, Pittock SJ, Britton JW, Hoye SL, Quek AM, et al.
MRI findings in autoimmune voltage-gated potassium channel
complex encephalitis with seizures: one potential etiology
for mesial temporal sclerosis. Am J Neuroradiol.
2014;35:84-9.
40. Schmitt SE, Pargeon
K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D, et al.
Extreme delta brush: a unique EEG pattern in adults with
anti-NMDA receptor encephalitis. Neurology. 2012;
79:1094-100.
41. Hacohen Y, Singh R,
Rossi M, Lang B, Hemingway C, Lim M, et al. Clinical
relevance of voltage-gated potassium channel-complex
antibodies in children. Neurology. 2015;85:967-75.
42. Dalmau J, Gleichman
AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al.
Anti-NMDA-receptor encephalitis: Case series and analysis of
the effects of antibodies. Lancet Neurol. 2008;7:1091-8.
43. Gresa-Arribas N,
Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F,
et al. Antibody titres at diagnosis and during
follow-up of anti-NMDA receptor encephalitis: A
retrospective study. Lancet Neurol. 2014;13:167-77.
44. Nosadini M,
Mohammad SS, Ramanathan S, Brilot F, Dale RC. Immune therapy
in autoimmune encephalitis: a systematic review. Expert Rev
Neurother. 2015;15:
1391-419.
45. Suppiej A, Nosadini
M, Zuliani L, Pelizza MF, Toldo I, Bertossi C, et al.
Plasma exchange in pediatric anti-NMDAR encephalitis: a
systematic review. Brain Dev. 2016;38:613-22.
46. Nagappa M, Bindu
PS, Mahadevan A, Sinha S, Mathuranath PS, Taly AB. Clinical
features, therapeutic response, and follow-up in pediatric
Anti-N-Methyl-D-Aspartate Receptor encephalitis: Experience
from a tertiary care university hospital in India.
Neuropediatrics. 2016;47:24-32.
47. Bar-Or A, Fawaz L,
Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al.
Abnormal B-cell cytokine responses a trigger of
T-cell-mediated disease in MS? Ann Neurol. 2010;67:452-61.
48. Nosadini M, Alper
G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S, et al.
Rituximab monitoring and redosing in pediatric neuromyelitis
optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm.
2016;3: e188.
49.
Dale RC, Brilot F, Duffy LV, Twilt M, Waldman AT,
Narula S, Muscal E, et al. Utility and safety of
rituximab in pediatric autoimmune and inflammatory CNS
disease. Neurology. 2014; 83:142-50.
50. Behrendt V, Krogias
C, Reinacher-Schick A, Gold R, Kleiter I, et al.
Bortezomib treatment for patients with
anti-N-methyl-D-aspartate receptor encephalitis. JAMA
Neurol. 2016;73:1251-53.
51. Lee WJ, Lee ST,
Moon J, Sunwoo JS, Byun JI, Lim JA, et al.
Tocilizumab in autoimmune encephalitis refractory to
rituximab: An institutional cohort study.
Neurothera-peutics. 2016;13:824-32.
52. Kothur K, Wienholt
L, Mohammad SS, Tantsis EM, Pillai S, Britton PN, et al.
Utility of CSF cytokine/chemokines as markers of active
intrathecal inflammation: comparison of demyelinating,
anti-NMDAR and enteroviral encephalitis. PLoS One.
2016;11:e0161656.
53. Tatencloux S,
Chretien P, Rogemond V, Honnorat J, Tardieu M, Deiva K.
Intrathecal treatment of anti-N- Methyl-D-aspartate receptor
encephalitis in children. Dev Med Child Neurol.
2015;57:95-9.
54. Nosadini M,
Mohammad SS, Toldo I, Sartori S, Dale RC. Mycophenolate
mofetil, azathioprine and methotrexate usage in paediatric
anti-NMDAR encephalitis: A systematic literature review. Eur
J Paediatr Neurol. 2019;23:7-18.



