|
|
|
Indian Pediatr 2019;56: 603-605 |
 |
Vici Syndrome with a
Novel Mutation in EPG5
|
|
Amita Moirangthem 1,
Kausik Mandal1,
Apurba Ghosh2 and
Shubha R Phadke1
From 1Department of Medical Genetics,
Sanjay Gandhi Postgraduate Institute of Medical Sciences (SGPGIMS),
Lucknow, Uttar Pradesh, India, and 2Institute of Child
Health, Kolkata, West Bengal, India
Correspondence to: Dr Shubha R Phadke, Professor and
Head, Department of Medical Genetics, Sanjay Gandhi Postgraduate
Institute of Medical Sciences, Lucknow 226 014, Uttar Pradesh, India.
Email:
[email protected]
Received: October 31, 2018;
Initial review: February 25, 2019;
Accepted: May 14, 2019.
|
Background: Vici syndrome is a neurodevelopmental
disorder of the autophagy pathway. Almost all cases reported have the
cardinal features of agenesis of corpus callosum, cataract,
cardiomyopathy, immunodeficiency and hypopigmentation. Case
characteristics: 8-month-old boy with developmental delay, myoclonic
jerks, repeated respiratory infections, coarse facial features, cataract
and hypopigmented hair. Echocardiography revealed dilated cardiomyopathy
and magnetic resonance imaging of brain suggested agenesis of corpus
callosum. Exome sequencing detected a novel homozygous nonsense mutation
in the EPG5 gene. Outcome: Establishing a definite
diagnosis helped in proper prognostication, providing genetic counseling
and prenatal diagnosis to the family. Message: Though uncommon,
presence of the characteristic features makes Vici syndrome a clinically
recognizable cause of developmental delay.
Keywords: Agenesis of corpus callosum, Autophagy, Cataract,
Developmental delay, Hypopigmentation.
|
|
V
ici syndrome (OMIM#24280) is a multisystem
disorder of the autophagy pathway. The typical phenotype includes severe
developmental delay, agenesis of corpus callosum, cardiomyopathy,
cataract, generalized hypopigmentation and variable immunodeficiency.
Additional features like coarse facies and gingival hyperplasia makes
this rare disease a close differential diagnosis of lysosomal storage
disorders. Bi-allelic mutation in the ectopic P-granules autophagy
protein 5, (EPG5), is the underlying etiology. We describe
the disease in an infant, with history of similarly affected siblings,
and report a novel homozygous mutation in EPG5 in the proband.
Case Report
The propositus presented at eight months of age with
severe global developmental delay and myoclonic jerks. He had total head
lag and no social smile. He also had repeated respiratory tract
infections and was hospitalized thrice for pneumonia. He was born at 37
weeks with a birth weight of 2.75 kg and had an uneventful perinatal
course. His parents were second cousins once-removed, and he had a
deceased elder brother who had similar features.
The proband had a weight of 7 kg (-2 SD), length of
67 cm (-1 to -2 SD) and occipito-frontal circumference of 40 cm (-3 to
-4 SD). He had coarse facial features, prominent metopic suture,
bitemporal narrowing, frontal upsweep of hair, arched well-defined
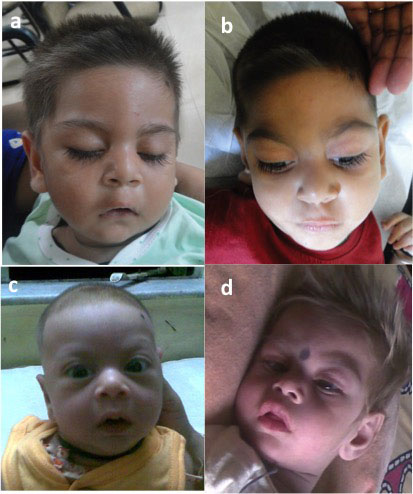
eyebrows, smooth philtrum, tented upper lip and retrognathia (Fig.
1a). Gingival hyperplasia and cataract in both eyes were also noted.
Hair was lighter in color as compared to the parents. He also had
tapering fingers, left single palmar crease and brachydactyly,
especially of the feet. The liver was palpable 2 cm below right costal
margin (span of 5 cm) and firm in consistency. Spleen was not palpable.
He did not have any eye contact and responded only to loud noise. There
was generalized hypotonia and deep tendon reflexes were not elicitable,
but there was no focal neurological deficit.
His blood investigations at various instances showed
hemoglobin of 9.6 -10.3 g/dL, platelet count 525-684 x 10 9/L,
total leucocyte count 21.6-25.3 x 109/L
with 33-46% neutrophils and 48-58% lymphocytes. The general blood
picture was normocytic normochromic with occasional hypochromic
microcytic cells. Creatine phosphokinase was raised (358 U/L; normal
<190 U/L). Random blood glucose, liver- and renal-function tests were
unremarkable. Thyroid profile was normal. There was elevated C-reactive
protein (43.6 mg/dL). Tandem mass spectroscopy did not reveal any
abnormal metabolite. Serum antibody levels and other immunological
work-up could not be performed.
Eye examination showed anterior polar cataract, disc
pallor and abnormal visual evoked potentials (VEP) in both eyes.
Echocardiography showed features of dilated cardiomyopathy; global left
ventricular hypokinesia (ejection fraction 44%) and mild mitral
regurgitation. Corpus callosal agenesis with dorsal cyst and mild lag in
myelination were noted in magnetic resonance imaging (MRI) of brain (Web
Fig. 1). Skeletal radiographs did not show any feature of
dysostosis multiplex and urine thin layer chromatography for lysosomal
storage disorders was also normal. Enzyme assay for GM1 gangliosidosis,
mucopolysaccharidosis type I and mucolipidosis were within normal
limits.
Genomic DNA was extracted from the blood of the
patient and parents. The libraries were prepared with the NexteraRapid
Capture Exome, Illumina and sequenced on HiSeq 4000 platform (Illumina,
San Diego, CA, USA). Sequences were aligned to the GRCh37/hg19 human
reference genome. The average coverage in the proband was 176x and 97.9
% of the exome was covered at >20x. Bioinformatics analysis identified a
homozygous novel variant c.3544G>T (p.Glu1182*) in EPG5
(NM_020964.2) in the proband. Both parents were heterozygous carriers
for the same. Validation of the variant by Sanger sequencing was done in
the proband and his parents using ABI 310 capillary sequencer (Applied
Biosystems, Foster City, CA, USA).
The proband was again examined at three years of age.
He had achieved neck control but could not roll over or sit with
support. He did not have any meaningful speech. He had feeding
difficulty and had several episodes of respiratory tract infections
requiring hospital admission. Hypopigmentation was mild. His facial
features had coarsened further (Fig. 1b). Generalized
hypotonia persisted but with intermittent episodes of spasticity. He had
developed joint contractures of the elbows, wrists, knees, ankles and
small joints of the hands.
 |
|
Fig. 1 Proband at the ages of 8
months (a); and 3 years (b); proband’s elder brother at 3 months
(c), and 2 years (d).
|
The elder brother of the proband was also similarly
affected and expired at 2 years 3 months. He was not examined personally
but his medical records and photographs were reviewed. He was born at 38
completed weeks with a birth weight of 2.7 kg. He had feeding
difficulty, hypotonia during the neonatal period. Epicanthal folds, high
arched palate and retrognathia were noted. He had severe developmental
delay, myoclonic jerks and never achieved head control and social smile.
He had recurrent pneumonia and succumbed during such an episode.
Photographs showed similar coarseness of facies as the proband (Fig.
1 c,d). He had obvious generalized hypopigmentation. Dysgenesis of
corpus callosum and periventricular white matter hypoplasia were noted
in MRI brain. VEP was abnormal in both eyes. Biotinidase assay and
multiplex PCR for spinal muscular atrophy (SMA) were normal. His tandem
mass spectroscopy was unremarkable. His sample was not available for
further genetic testing.
Prenatal diagnosis could be provided to the family in
the third pregnancy. The mutation was not detected in the fetus and a
healthy child was born.
Discussion
Though several cases of Vici syndrome were reported
after it was first described in 1988, the causative gene EPG5 at
18q12.3 was identified by exome sequencing in 2012 [1]. The largest
study of Vici syndrome described 50 patients from 30 families. The
cardinal features of developmental delay, agenesis of corpus callosum,
cataract, cardiomyopathy, hypopigmentation and skeletal myopathy were
present in all cases [2,3]. Inconsistent presence of these features was
reported in a recent study from Japan [4].
In the present family, there was some degree of
phenotypic variability in the affected siblings. The elder sibling had
more profound developmental delay. He also had more severe
hypopigmentation which became more obvious with age as opposed to the
proband. The arched and well delineated eyebrows, prominent metopic
suture and upsweep of hair giving a characteristic facial phenotype in
the proband were not conspicuous in the photographs of the sibling, and
also have not been reported previously. In addition, our case had
leukocytosis in contrast to the more commonly observed finding of
leucopenia [3].
This disorder is one of the emerging group of
metabolic disorders known as congenital disorders of autophagy [5].
EPG5 encodes a protein with an important role in autophagy network
which explains the multi-systemic involvement and overlapping features
of lysosomal storage disorders. Defective autophagosome-lysosomal fusion
has been demonstrated in mice models and more recently in cultured skin
fibroblasts of patients [4]. Majority of the mutations reported so far
are truncating mutations as also observed in our case. These are
distributed throughout the 44 exons and splice sites [1-3,6-8]. Our
patient had a nonsense substitution, c.3544G>T (p.Glu1182*) in the 25 th
exon leading to a premature termination codon. This variant has not been
observed in Exac (exac.broadinstitute.org), gnomAD (gnomad.broadinstitute.org)
and 1000 Genomes Project (internationalgenome.org). It occurs at
an aminoacid that is conserved across species and is predicted to be
disease-causing by in-silico analysis (mutation-taster.org).
This report adds to the pan-ethnic occurrence of Vici
syndrome and highlights intra-familial variability. With more widespread
use of next generation sequencing more cases of this under-diagnosed
condition could be identified including cases with atypical phenotype
and fetal manifestations [9].
Contributors: AM: drafting the manuscript,
compilation of clinical details of the patient, analysis and
interpretation of exome data; KM: procuring patient details and
management of the family, and critical inputs to the manuscript; AG:
clinical management of the patient; SRP: critical revision and final
approval. All authors approved the final version of manuscript and agree
to be accountable for authenticity and integrity of the work.
Funding: None; Competing interest: None
stated.
Consent: Written informed consent was taken from
the family for publication of clinical details and photographs of the
patients.
References
1. Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B,
Smith F, Urry Z, et al. Recessive mutations in EPG5 cause Vici
syndrome, a multisystem disorder with defective autophagy. Nat Genet.
2013;45:83-7.
2. Byrne S, Dionisi-Vici C, Smith L, Gautel M,
Jungbluth H. Vici syndrome: A review. Orphanet J Rare Dis. 2016;11:21.
3. Byrne S, Jansen L, U-King-Im JM, Siddiqui A, Lidov
HG, Bodi I, et al. EPG5-related Vici syndrome: A paradigm
of neurodevelopmental disorders with defective autophagy. Brain.
2016;139:765-81.
4. Hori I, Otomo T, Nakashima M, Miya F, Negishi Y,
Shiraishi H, et al. Defects in autophagosome-lysosome fusion
underlie Vici syndrome, a neurodevelopmental disorder with multisystem
involvement. Sci Rep. 2017;7:3552.
5. Ebrahimi-Fakhari D, Saffari A, Wahlster L, Lu J,
Byrne S, Hoffmann GF, et al. Congenital disorders of autophagy:
an emerging novel class of inborn errors of neuro-metabolism. Brain.
2016;139:317-37.
6. Tasdemir S, Sahin I, Cayýr A, Yuce I, Ceylaner S,
Tatar A. Vici syndrome in siblings born to consanguineous parents. Am J
Med Genet A. 2016;170A:220-5.
7. Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi
P, Mehdizadeh M, et al. First description of a patient with Vici
syndrome due to a mutation affecting the penultimate exon of EPG5
and review of the literature. Am J Med Genet A. 2014;164A:3170-5.
8. Kane MS, Vilboux T, Wolfe LA, Lee PR, Wang Y,
Huddleston KC, et al. Aberrant splicing induced by the most
common EPG5 mutation in an individual with Vici syndrome. Brain.
2016;139:1-4.
9. Aggarwal S, Tandon A, Bhowmik AD, Dalal A.
Autopsy findings in EPG5-related Vici syndrome with antenatal onset:
Additional report of Focal cortical microdysgenesis in a second
trimester fetus. Am J Med Genet A. 2018;176:499-501.
|
|
|
 |
|
