|
|
|
Indian Pediatr 2019;56: 556-557 |
 |
Molecular and Histopathological
Characterization of Patients Presenting with the Duchenne
Muscular Dystrophy Phenotype in a Tertiary Care Center in
Southern India
|
|
Karthik Tallapaka 1,2,
Prajnya Ranganath1,2,
Angalena Ramachandran2,
Megha S Uppin3, Sreeja
Perala1,2, Shagun Aggarwal1,2,
Dhanya Lakshmi1,2, AK Meena4
and Ashwin B Dalal2
From Departments of 1Medical Genetics, 3Pathology,
4Neurology, Nizam’s Institute of Medical Sciences & 2Diagnostics
division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad,
Telangana, India
Correspondence to: Dr Prajnya Ranganath, Associate
Professor and Head, Department of Medical Genetics, Nizam’s Institute of
Medical Sciences, Panjagutta, Hyderabad, Telangana 500 082, India.
prajnyaranganath@gmail.com
Received: July 26, 2018;
Initial review: December 17, 2018;
Accepted: May 13, 2019.
|
|
Objective: To study the
histopathological characteristics and mutation spectrum of patients
presenting with the Duchenne muscular dystrophy (DMD) phenotype.
Methods: This was a descriptive study conducted over a period of 8
years. Multiplex ligation-dependent probe amplification (MLPA) was done
in patients presenting with the DMD phenotype. If MLPA was negative,
patients were offered muscle biopsy for histopathological studies and/or
next generation sequencing (NGS) based multigene panel testing for
muscular dystrophies. Results: Of the 510 patients included,
mutation in the DMD gene was detected by MLPA in 372 (72.9%), of
whom 342 (67.1%) had exonic deletions and 30 (5.9%) had exonic
duplications. Exons 45-55 were most commonly involved in large deletions
and exons 1-10 were the commonest exons involved in duplications. In the
MLPA-negative cohort, 27 proceeded for muscle biopsy. NGS was done in 14
patients, 10 of whom had pathogenic mutations in the DMD gene, 3
were non dystrophinopathies and no pathogenic variant could be
identified in one patient. Conclusions: For patients presenting
with the DMD phenotype, MLPA of the DMD gene has a high
diagnostic rate of about 73%, and non-dystrophinopathies may constitute
a small but significant proportion.
Keywords: Diagnosis, Histopathology, Multiplex
ligation-dependent probe amplification, Neuromuscular disorders, Next
generation sequencing.
|
|
W
ith an incidence of 1 in 3500, Duchenne muscular
dystrophy (DMD) is the commonest genetically inherited primary muscle
disease [1]. DMD is an X-linked condition caused by mutation in the
DMD gene which codes for dystrophin, and usually presents in boys
within the first decade of life with rapidly progressive proximal muscle
weakness. Limb girdle muscle dystrophies (LGMD) are a group of disorders
mostly inherited in an autosomal recessive or dominant fashion, some of
which, especially the sarcoglycanopathies, can clinically resemble DMD.
Multiplex PCR (mPCR) was the preferred first-line
genetic test for DMD until the advent of Multiplex Ligation-dependent
Probe Amplification (MLPA), which in comparison to mPCR, has better
sensitivity in detecting deletions and can detect duplications and
carrier status as well. However, point mutations and small indels are
not detected by MLPA. With the advent of next generation sequencing
(NGS), these sequence variants are being more frequently identified.
This study describes the diagnostic yield of MLPA and the spectrum of
mutations (including point mutations) occurring in DMD patients hailing
from Southern India. In this era of rapid technological advancement, it
is pertinent that the mutation pattern be known so that specific
treatment options could be offered with the likely availability of
mutation-specific therapies in the not-so-distant future.
Methods
This observational study was conducted between March
2009 and June 2017 at a tertiary care hospital in Southern India. After
the approval of the Institute Ethics committee, informed consent was
obtained from parents of patients with the DMD phenotype. Boys aged 2 to
18 years were considered to have the DMD phenotype if one or more
of the following features were present: history of progressive,
symmetrical muscle weakness with onset in early childhood; history of
motor developmental delay; positive family history suggestive of
X-linked inheritance; weakness predominantly in the proximal muscles;
earlier onset and more severe weakness in the lower limbs compared to
the upper limbs with positive Gower’s sign; calf muscle hypertrophy;
raised serum creatine phosphokinase (CPK) to more than 10 times the
normal value; and loss of independent ambulation by the age of 13 years.
Detailed clinical features and the results of
investigations like serum creatine phosphokinase (CPK), electromyography
(EMG) and muscle biopsy if already performed, were recorded. MLPA (MRC,
Holland) was done as per the manufacturer’s protocol to detect deletions
and duplications of the DMD gene. As per the Institute protocol,
if the MLPA test results were negative, the patients were offered muscle
biopsy for histopathological studies. From the year 2014, NGS-based
multigene panel testing for muscular dystrophies was offered as the
other option. The patients were counseled regarding the advantages and
disadvantages of both the choices. In the first choice after the
confirmation of muscular dystrophy on histopathology,
immuno-histochemistry (IHC) of the fresh frozen biopsy specimen was
offered with staining for dystrophin 1, 2 and 3 (Novocastra –
lyophilised mouse monoclonal antibodies by Leica biosystems). If
negative, then IHC staining for alpha, beta, gamma and delta
sarcoglycans & dysferlin (Leica biosystems) was advised. NGS was
outsourced to a commercial diagnostic lab, if the family opted for it.
Results
A total of 510 patients were included in the study.
The median (range) age of presentation was 8 (3-10) years. All the
patients included in the study had proximal muscle weakness in the lower
limbs at the time of presentation. Calf hypertrophy, developmental delay
(motor/global) and raised serum transaminase levels were the other
reasons for consultation. Family history was positive for DMD in 25% of
the cases. Global developmental delay or poor scholastic performance was
noted in about 26% of the children. The median (range) serum CPK level
was 10,000 (5762-14960) IU. 2D echocardiography showed a mildly dilated
left heart with preserved ejection fraction in only one patient aged 10
years, but none of the other patients had features of cardiac
insufficiency in the initial cardiac evaluation done at the time of
diagnosis.
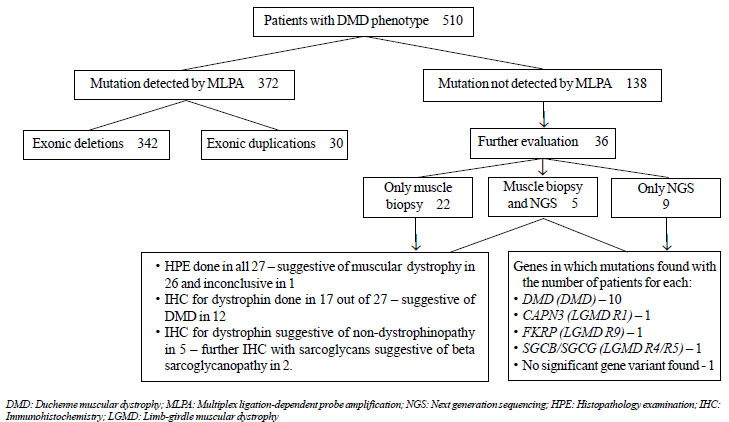 |
|
Fig. 1 Flowchart summarizing the
histopathological and molecular genetic evaluation of the
patients in the present study.
|
The results of the histopathological and molecular
genetic evaluation are summarized in the flowchart in Fig. 1.
Of the 510 patients, mutation in the DMD gene was detected by
MLPA in 372 (72.9%). Of the mutations identified, 342 (67.1%) were
exonic deletions and the remaining 30 (5.9%) were exonic duplications.
Exons 45-55 were most commonly involved in large deletions [245 (71.64%)
of the deletion mutations]. Exons 1-10 were the commonest exons involved
in duplications [17 (56.67%) of the duplication mutations]. Only a
single exon was involved in either deletion or duplication in 72
(19.35%) patients, with exon 45 deletion being the most common single
exon deletion. One child with deletion of exon 1 presented with a
slightly milder phenotype, probably due to alternative initiation of
translation [2]. One patient had a complex deletion involving exon 1 and
exons 44-49.
Thirty-six out of the total 138 MLPA-negative
patients underwent further evaluation through muscle biopsy [22 (15.9%)]
or NGS [9 (6.5%)] or both [5 (3.6%)]. The remaining patients [102
(73.9%)] did not opt to undergo further evaluation due to financial
constraints, or were lost to follow-up.
Out of the 27 MLPA-negative patients who opted for
muscle biopsy, IHC for dystrophin could be done in only 17 patients.
Twelve of them showed absence of dystrophin staining along the
sarcolemma, suggestive of dystrophinopathy. The remaining five showed
presence of normal dystrophin staining which ruled out dystrophinopathy.
Out of these five cases, further IHC with alpha, beta, gamma and delta
sarcoglycans showed a pattern suggestive of beta sarcoglycanopathy in
two cases and in the other three further characterization could not be
done. Representative histopathological pictures are shown in
Web
Fig 1. Five of the 12 patients whose dystrophin immunostaining
was suggestive of dystrophinopathy opted for further NGS testing also,
in order to identify the exact disease-causing mutation, either for the
purpose of prenatal diagnosis and/or carrier testing of family members
through targeted mutation analysis, or with the intent of ascertaining
suitability for mutation-specific therapies such as stop codon
read-through therapy.
NGS was done in a total of 14 MLPA-negative patients;
following confirmation of dystrophinopathy through muscle biopsy and IHC
in 5 and directly after MLPA in 9. Out of them, 10 had sequence variants
in the DMD gene (transcript id ENST00000357033), with known
mutations in 5 (c.6283C>T, c.8692C>T, c.3257dupA, c.10454_10454delT and
c.2047G>T) and novel variants predicted to be pathogenic in 5
(c.2168_2168+1delGGinsCT,c.5478 _5490del TCTTTGCAACAAT, c.3154A>T,c.
3037G>T and c.2646_2646delT). Three patients were identified to have
non-dystrophinopathic muscular dystrophy through NGS – one with limb
girdle muscular dystrophy autosomal recessive 1 (LGMDR1/ LGMD2A/
Calpainopathy; homozygous known mutation c.802-9G>A in CAPN3
gene); one with LGMD autosomal recessive 9 (LGMDR9/ LGMD2I/ LGMD-Dystro-glycanopathy
type C5; homozygous known mutation c.1343C>T in FKRP gene); and
one with variants of uncertain significance – homozygous c. 799C>T
variant in SGCB and compound heterozygous variants
c.479T>C and c.653T>C in SGCG (LGMD R4 or R5/ beta or gamma
sarcoglycanopathy). In the last patient, further muscle biopsy and IHC
with beta and gamma sarcoglycans was recommended for further functional
corroboration and characterization, but the family did not opt for it.
NGS did not reveal any significant gene variants in one patient.
Discussion
A large proportion of cases of DMD are caused by
large deletions/duplications. Initial Indian studies which were largely
based on mPCR, reported a diagnostic rate of about 60-70% for this
diagnostic modality [3-6]. Verma, et al. [7] found that MLPA had
an additional diagnostic yield of 5.6% when compared to mPCR. The
diagnostic rate of MLPA in our cohort was around 73%. Majority of the
deletion mutations identified in our study involved the hot spot region
in the central domain between exons 45-55, which is consistent with what
is reported in many previous studies [3-6]. Duplications on the other
hand were found to be commoner in the initial 10 exons. These exonic
regions are frequently involved in duplication mutations, in various
populations across the world [8].
Next generation sequencing-based muscular dystrophy
gene panel sequencing was successful in identifying the mutation in all
but one case where it could be done. A few recently published studies
have demonstrated the successful usage of NGS for detection of large
deletions and duplications also and thus this could become the
investigation of choice for DMD in the near future [9]. However, as can
be seen in our study, NGS was feasible only in 14 patients out of the
138 MLPA-negative cases who needed it. In spite of a significant
decrease in the price of NGS in the past few years, the cost still
remains a major deterrent for many families from the lower socioeconomic
strata.
Muscle biopsy (histopathology and
immuno-histochemistry) may not be preferable as the first line
investigation in the diagnosis of MLPA-negative patients due to its
invasive nature, but is helpful in resolving variants of uncertain
significance (VUS) identified by NGS. Skin biopsy has been shown in some
studies to be a sensitive and specific, less-invasive testing modality
for dystrophinopathies and could be considered as an alternative option
for histopathological characterization of MLPA-negative patients [10].
Seventy-two patients had a mutation which is amenable
to the recently approved exon 51 skipping therapy (Eteplirsen) and
forty-six others had mutations which are potentially amenable to exon 53
skipping. Five patients with nonsense mutations who may potentially
benefit with stop codon read-through therapy have also been identified.
Following initial characterisation of the DMD
phenotype as per the inclusion criteria of the study, further phenotypic
delineation was not done for the patients. Therefore, genotype-phenotype
correlation could not be established for the different types of
mutations identified, which is one of the limitations of this study. As
an extension of this study, further deep phenotyping of the patients is
planned with correlation of the type of mutation with the age at onset,
severity and/or progression of individual phenotypic parameters.
To conclude, MLPA has a good diagnostic rate for DMD
and should be the first line genetic investigation of choice in a child
presenting with the DMD phenotype. Non-dystrophinopathies, especially
the childhood-onset autosomal recessive limb girdle muscular
dystrophies, may constitute a small but significant proportion of
patients presenting with the DMD phenotype, who test negative by MLPA.
Next generation sequencing-based multigene panel testing for muscular
dystrophy-associated genes, because of its non-invasive nature and its
power to identify mutations in various DMD mimickers, should be offered
to all MLPA-negative cases with the DMD phenotype. Identification of the
exact disease-causing mutation(s) would help not just in molecular
confirmation of the diagnosis, but also in appropriate counseling and in
offering prenatal testing and carrier screening for the families. It
would also be of help in identifying patients amenable to the various
mutation-specific therapies that are being developed and/or
investigated. However, cost is still a deterrent for doing NGS-based
molecular studies in our country, especially in resource-poor settings.
Thus, there is a need to devise strategies to lower the cost of
diagnostic work up in MLPA-negative cases.
Acknowledgements: MedGenome Labs Private
Ltd., Bengaluru, India for performing next generation sequencing-based
molecular genetic studies.
Contributors: KT: design and
conceptualization of the study, clinical evaluation, molecular analysis,
collection of data, preparation of manuscript; PR: design and
conceptualization of the study, clinical evaluation, analysis of data
and preparation of manuscript; AR: molecular analysis (MLPA); MSU:
muscle biopsy and histopathology; SP: collection of data; SA, DLN, AKM:
clinical evaluation of patients; ABD: clinical evaluation, molecular
analysis and critical review of manuscript.
Funding: None; Competing interest: None
stated.
|
What This Study Adds?
•
MLPA of the DMD gene has
a high (73%) diagnostic rate in patients with the DMD phenotype.
•
Next generation sequencing, a non-invasive and precise
diagnostic modality, has the potential to replace invasive
techniques such as muscle biopsy as the preferred investigation
for MLPA-negative DMD patients.
|
References
1. Emery AE. Population frequencies of inherited
neuromuscular diseases: A world survey. Neuromuscul Disord.
1991;1:19-29.
2. Gurvich OL, Maiti B, Weiss RB, Aggarwal G, Howard
MT, Flanigan KM. DMD exon 1 truncating point mutations: amelioration of
phenotype by alternative translation initiation in exon 6. Hum Mutat.
2009;30:633-40.
3. Swaminathan B, Shubha GN, Shubha D, Murthy AR,
Kiran Kumar HB, Shylashree S, et al. Duchenne muscular dystrophy:
a clinical, histopathological and genetic study at a neurology tertiary
care center in Southern India. Neurol India. 2009;57:734-8.
4. Banerjee M, Verma IC. Are there ethnic differences
in deletions in the dystrophin gene? Am J Med Genet. 1997;68:152-7.
5. Basumatary LJ, Das M, Goswami M, Kayal AK.
Deletion pattern in the dystrophin gene in Duchenne muscular dystrophy
patients in northeast India. J Neurosci Rural Pract. 2013;4:227-9.
6. Singh V, Sinha S, Mishra S, Chaturvedi LS, Pradhan
S, Mittal RD, et al. Proportion and pattern of dystrophin gene
deletions in north Indian Duchenne and Becker muscular dystrophy
patients. Hum Genet. 1997;99:206-8.
7. Verma PK, Dalal A, Mittal B, Phadke SR. Utility of
MLPA in mutation analysis and carrier detection for Duchenne muscular
dystrophy. Indian J Hum Genet. 2012;18:91-4.
8. Bladen CL, Salgado D, Monges S, Foncuberta ME,
Kekou K, Kosma K, et al. The TREAT-NMD DMD Global Database:
analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum
Mutat. 2015;36:395-402.
9. Okubo M, Minami N, Goto K, Goto Y, Noguchi S,
Mitsuhashi S, et al. Genetic diagnosis of Duchenne/Becker
muscular dystrophy using next-generation sequencing: validation analysis
of DMD mutations. J Hum Genet. 2016;61:483-9.
10. Chakrabarty B, Sharma MC, Gulati S, Kabra M,
Pandey RM, Sarkar C. Dystrophinopathy diagnosis made easy: Skin biopsy,
an emerging novel tool. J Child Neurol. 2014;29:469-74.
|
|
|
 |
|
