|
Clinicopathological Conference |
|
|
Indian Pediatr 2015;52: 601-606 |
 |
An Infant with Prolonged Fever
|
|
*Kirti Gupta, Deepti Suri, Avinash Sharma,
Amit Rawat and Surjit Singh
From *Department of Histopathology; and Pediatric
Allergy-Immunology Unit, Department of Pediatrics; PGIMER, Chandigarh.
India.
Correspondence to: Dr Kirti Gupta, Additional
Professor, Department of Histopathology, Postgraduate Institute of
Medical Education and Research (PGIMER), Chandigarh, India.
Email:
kirtigupta10@yahoo.co.in
|
|
W
e present an infant who presented with fever and
generalized rash. She also had hepatosplenomegaly and persistent
respiratory distress, and later developed multi-organ failure.
Clinical Protocol
History: A seven-month-old, developmentally
normal girl – born at term with birth weight of 2.5 kg to a
non-consanguineously married couple – presented with fever and rash for
1½ months. The illness started with generalized erythematous,
maculopapular rash involving the flexural areas, scalp and trunk, along
with intermittent fever. The rash subsided over 15 days leaving behind a
dry, scaly skin. During this period, she also developed cough and rapid
breathing, and had received intravenous antimicrobials elsewhere with
only partial response. The child was referred to our institute in view
of reappearance of fever, rash and pedal edema. At admission, she had
fever, cough, rapid breathing, poor feeding and irritability, along with
dry scaly skin and fading rash. The child was first in birth order with
no significant family history of prolonged illness, tuberculosis, or any
unexplained death in family. She was immunized for age, and BCG site had
a normal scar.
Clinical examination: She weighed 5.6 kg (<–2
SD); length was 62 cm (-2 SD). She was afebrile with pulse rate of
106/min, respiratory rate of 56/min and blood pressure of 94/60 mmHg.
The skin was dry, scaly and eczematous with rash predominantly over the
scalp and lower limbs with involvement of palms and soles. Bones and
joints were normal as was examination of ears, nose and throat. Systemic
examination revealed soft, distended abdomen with palpable liver (5 cm
under costal margin with a span of 7 cm) and spleen (1.5 cm under left
costal margin). The child also had tachypnea along with subcostal and
substernal retractions; crepitations were auscultable over bilateral
lung fields. Cardiovascular and neurological examinations were
unremarkable.
Investigations (Tables I and
II): Before presenting to our institute: C reactive protein (CRP)
1.2 mg/dL, lactate dehydrogenase (LDH) 246, procalcitonin 0.3 ng/mL, S.
ferritin <10 ng/mL, ultrasonography abdomen normal, 2D-echocardiography
(ECHO) and chest X-ray (CXR) normal, HIV serology non-reactive,
hepatitis B surface antigen (HBsAg) negative. Dengue serology, malaria
serology and widal tests were also negative. Blood culture grew
Pseudomonas stutzeri and Acinetobacter woffii. Immunoglobulin
levels were: IgA 87 mg/dL and IgG 1176 mg/dL.
TABLE I Hematological Investigations in the Present Case
|
Date |
4/08/14 |
27/08/14 |
17/09/14 |
30/09/14 |
7/10/14 |
|
Hb (g/dl) |
7.7 |
9.2 |
7.0 |
10.2 |
8.0 |
|
TLC/mm3 |
23,900 |
24,900 |
23,600 |
14,700 |
50,300 |
|
DLC (%) |
P56 L37 |
P50L47 |
P55L39M3E3 |
P39L49M9E3 |
P80L10M3E1 |
|
ANC/mm3 |
13,384 |
12,450 |
12,980 |
5,733 |
40,000 |
|
ALC/mm3 |
8,843 |
11,703 |
9,204 |
7,203 |
5,030 |
|
Platelets/mm3 |
5.20 |
7.49 |
7.87 |
6.98 |
4.26 |
|
ESR (westergreen, mm in 1st hr) |
44 |
51 |
24 |
7 |
|
TABLE II Biochemical Investigations in Present Case
|
Date |
24/09 |
04/10 |
13/10 |
|
Sodium |
137 |
138 |
141 |
|
Potassium |
5.7 |
6.0 |
5.3 |
|
Ca/PO4 |
9.2/4.9 |
9.8/4.6 |
8.2/3.4 |
|
Urea |
18 |
22 |
57 |
|
Creatinine |
0.40.4 |
0.6 |
0.8 |
|
S.Bilirubin |
0.7 |
0.7 |
|
|
S.Protein/Albumin |
5.5/NA |
6.4/2.4 |
|
|
SGOT/SGPT |
28/NA |
56/33 |
|
|
Cholesterol |
|
117 |
|
|
Triglycerides |
|
557 |
|
|
CRP |
|
28 |
|
|
NA-Not available; Ca-serum calcium; Ph.-serum phosphorus;
CRP- C-reactive protein. |
Skin biopsy was done at our institute. Epidermis
showed acanthosis with hyperkeratosis. The dermis showed mild
perivascular infiltration with lympho-mononuclear cells and occasional
histiocytes. Investigations at our institute : S. ferritin:
6.9 ng/mL, HIV non-reactive; anti-nuclear antibodies and anti-nuclear
cytoplasmic antibodies were absent. Chest X-Ray and high
resolution CT scan chest revealed small focal area of consolidation in
right upper lobe. No evidence of bronchiectasis, pleural or pericardial
effusion were seen. Ultrasonography of abdomen showed mild hepatomegaly
with grade I fatty liver.
Bone marrow examination showed normocellular bone
marrow with normal hematopoietic elements and mild prominence of
histiocytes. CD1a staining was negative. Bronchoscopy was normal,
broncho-alveolar lavage (BAL) did not show acid-fast bacilli (AFB) or
fungus; BAL culture was sterile and BAL fluid did not have hemosiderin
laden macrophages.
Microbiological investigations: Blood culture and
sensitivity (thrice): sterile. Bone marrow culture: sterile,
Cytomegalovirus (CMV) IgM antibody: negative, CMV polymerase chain
reaction: negative, Mycoplasma serology: 1/64 (normal 1/32),
Toxoplasma serology, Brucella serology: negative; Gastric
lavage for AFB, fungus and Pneumocystis jiroveci were negative.
Endotracheal (ET) aspirate culture revealed growth of yeast; AFB stain
was negative. Urine culture was sterile and Galactomannan assay was
normal.
Immunological investigations: There was hyper-gammaglobulinemia
with elevated serum IgG [1294 mg/dL (N 300-900)], IgM [201 mg/dL (N
40-160)] and IgE [248 IU/mL (N 0-6.6)]. Nitro blue tetrazolium test
(NBT): Unstimulated 30%, stimulated 95%; Dihydrorhodamine (DHR): MFI
unstimulated 1688 (control 869); MFI stimulated 8886 (control 8211);
Oxidative Index: 5.2 (control 9.4), showed normal shift to right. Repeat
DHR 12 days later: oxidation after stimulation of neutrophils showed
minimal shift to right. MFI unstimulated 3180 (control 2622); MFI
stimulated 5367 (control 11,806). Oxidative index: 1.68 (control4.5). P 67phox:
reduced expression.
Course and management: The child received
antibiotics, but her respiratory distress worsened during hospital stay.
In view of persisting pneumonia, primary immuno-deficiency was
considered. Chronic granulo-matous disease (CGD) was considered as DHR
was abnormal and P 67phox
expression was low. Following bronchoscopy, respiratory distress
worsened, mechanical ventilation was initiated on 15th
day of hospital stay; vancomycin, piperacillin-tazobactam, amphotericin
and cotrimoxazole was started empirically. She was ventilated in SIMV
mode for 3 days followed by high frequency oscillatory ventilation for 8
days. She continued to worsen resulting in respiratory acidosis with
persistent hypoxemia. She received two red cell transfusions for anemia
during hospital stay, and later developed multi-organ dysfunction, renal
failure. She died on day 30 of hospital stay.
Unit’s final diagnosis: Chronic granulomatous
disease with severe pneumonia, health care associated sepsis, and acute
respiratory distress syndrome.
Discussion (Clinical discussant): This infant
presented with prolonged fever, and infections with an underlying immune
deficiency, inflammatory disorders or neoplastic disorders were
considered. The neoplastic disorders which can present in infancy with
prolonged fever are leukemia, lymphoma, Langerhans cell histiocytosis
(LCH), neuroblastoma, and Castleman’s disease. A normal bone marrow
examination excluded these diagnosis, except for LCH. Though skin biopsy
revealing a CD1a would have been more helpful, still in the absence of
bone disease, bone marrow showing CD1a negativity, and chest X-ray
not suggestive, LCH did not appear to be a clinical possibility. Among
the auto-inflammatory or autoimmune disorders, systemic lupus
erythematosus (SLE) could be ruled out as ANA was negative; absence of
gastrointestinal disease rules out the inflammatory bowel disease (IBD)
and absence of oral ulcers nearly rules out Behcet’s disease. Child did
not have features of Kawasaki disease, and normal cell counts and
ferritin levels made possibility of hemophagocytic-lympho-histiocytosis
unlikely. Systemic onset juvenile idiopathic arthritis (sJIA) may or may
not have arthritis as a prominent feature but presents classically as
fever, rash, hepatosplenomegaly and serositis. These patients have
polymorphonuclear leucocytosis and thrombocytosis which were seen in
this child. However, the rash is classically evanescent in sJIA, not
like fixed flexural rash as in this baby. The initial CRP and ESR were
normal, and a normal ferritin nearly rules out any inflammatory disorder
like sJIA.
Many infectious causes can lead to prolonged fever in
infancy. In this child, the focus was lung, and the organisms isolated
were Pseudomonas, Acinetobacter and Candida. With HIV being ruled
out, primary immune deficiencies should be considered. The conditions
which are likely to present at this age are; severe combined immune
deficiency (SCID), a hyper-IgM disorder, CGD and Wiskott Aldrich
syndrome (WAS). WAS is an X-linked disorder and is very unlikely in a
girl child. Hyper-IgM can have autosomal recessive or X-linked
transmission. Patients can have recurrent respiratory infections,
including Pneumocystis jiroveci pneumonia. They can also have
immune dysregulation and malignancies. Neutropenia may be seen and they
classically have low IgG, IgA and a high or normal IgM. The index child
had raised IgG along with IgM. CD40, which is usually low in autosomal
recessive hyper-IgM, was normal in this child, rendering the diagnosis
of hyper-IgM unlikely. SCID presents as severe fatal infections in
infancy and classically, these children have lymphopenia,
hypogammaglobulinemia and absent thymus. Therefore, SCID looks unlikely,
but European society for immunodeficiency (ESID) criteria for diagnosis
of SCID suggest laboratory criteria of low CD3 cells, and reduced naïve
CD4 and CD8 T cells. The index child having low CD3 and reduced naïve T
cells, SCID cannot be completely ruled out. These patients, because of
the autologous circulating T cells, frequently have skin rash, enlarged
lymph nodes and even normally sized thymus. They can present atypically
even beyond infancy with granulomas and lymphoid malignancies [1].Thus,
the index child could have had SCID without lymphopenia because of
autologous T cells or it could be a leaky SCID with hypomorphic
mutations which are not very lethal. The immunoglobulins in SCID can be
very variable because of transplacentally transferred IgG; in an event
of infection, the IgM can also rise [2]. SCID with maternal engraftment
can have T cells with no lymphopenia; they have a graft versus host
disease (GVHD) like illness, presenting with rash, hepatosplenomegaly
and raised IgE. These features were seen in this child; however, other
two important features of diarrhea and eosinophilia were not seen.
Chronic granulomatous disease is a result of
phagocyte dysfunction and patients present with recurrent bacterial deep
seated infections. Classical features of excessive inflammation(polymorphonuclear
leucocytosis, hypergammaglobulinemia) and an abnormal DHR were present
in this child. The P 67phox
expression was low, supporting the diagnosis of CGD. A child with CGD
usually has an abnormal NBT and an abnormal respiratory burst in
activated lymphocytes which is usually less than 5% of the control. In
this case, DHR was low but the difference from the control was not more
than 5%; and an NBT was normal. Situations in which one can have a
falsely abnormal DHR with normal NBTare: myeloperoxidase deficiency, the
SAPHO syndrome and G6PD deficiency. These are unlikely in the index
child, because these are reasonably milder immune deficiencies.
Therefore, autosomal recessive CGD (AR-CGD) appears to be a strong
possibility. AR-CGDs are milder diseases than X-linked CGD. They
have slow smouldering disease, leucocytosis, hypergammaglo-bulinemia,
abnormal DHR and can have low P67phox
expression. The low number of T cells and the naïve T cells can be
explained because of sepsis that can cause increase in apoptosis in both
the subsets of CD3 T cells, and can result in a falsely low CD3 T cells
[3].
The final diagnosis of the unit was pneumonia with
disseminated fungal disease, underlying primary immune deficiency, AR
CGD with severe sepsis and with low naïve T cells, a possibility of SCID
with maternal T cell engraftment.
Senior Resident of treating unit: The diagnosis
was difficult because initial NBT report was normal. However, with a
strong possibility of CGD, DHR was done which was abnormal following
which P 67phox was
assayed which helped clinch the diagnosis. Autosomal recessive CGD is
rare but in the presence of mutations in P67phox
genes, the diagnosis can be made. These children present with recurrent
infections and granulomatous inflammation.
Pediatrician 1: The crucial investigation here is
DHR. If the DHR is abnormal, diagnosis is almost certain. Though the DHR
shift is not as marked as one would see in X-linked CGD, which is the
commoner variant. However, with this DHR report, there really is no
differential diagnosis.
Chairman: It seems that this child was born with
some kind of immune deficiency and most likely CGD. Can other defects of
innate immune system like complement deficiencies present at this age
with bacterial infections, and do we need to rule them out in such
situations?
Clinical discussant: Yes, complement deficiencies
can also lead to recurrent infections but usually the children with
complement deficiencies present with recurrent Neisseria
infections and slow smouldering course not usually fatal in infancy.
Pathology Protocol
A partial autopsy was performed, and the peritoneal
cavity revealed ascites with 300 mL of straw-colored fluid. The pleural
cavities and pericardial cavity were normal. The lungs were heavy (150
g), and the overlying pleura was dull. Inspissated secretions were
identified within airways. Both lungs revealed lower lobe consolidation
with a firm texture and loss of crepitancy of lung parenchyma (Fig
1a). An infarct (1.5 cm), was identified in left lower lobe.
No areas of breaking-down abscesses or caseous necrotic foci were
detected. On histology, most of the respiratory and terminal bronchioles
revealed ulcerated epithelium and the lumen were filled with Periodic
acid-Schiff (PAS) positive inspissated secretions. The alveoli were
filled with pigmented macrophages (Fig 1b). The
pigment was PAS positive consistent with lipofuscin-like material. CD 68
immunostain highlighted these macrophages (Fig 1d).
Many multinucleated giant cells with ill-formed epithelioid granulomas
were noted within interstitium (Fig 1c). The rest
of the lung showed features of bronchopneumonia, diffuse alveolar
hemorrhage and occasional fibrin thrombi within pulmonary vein.
Additionally, the infarct microscopically revealed presence of
Aspergillus hyphae with early invasion into the lung parenchyma (Fig
1e). Gram’s stain, and stain for AFB and Nocardia were
negative. Polymerase chain reaction (PCR) for respiratory syncytial
virus, metapneumo virus, influenza virus and parainfluenza virus were
negative.
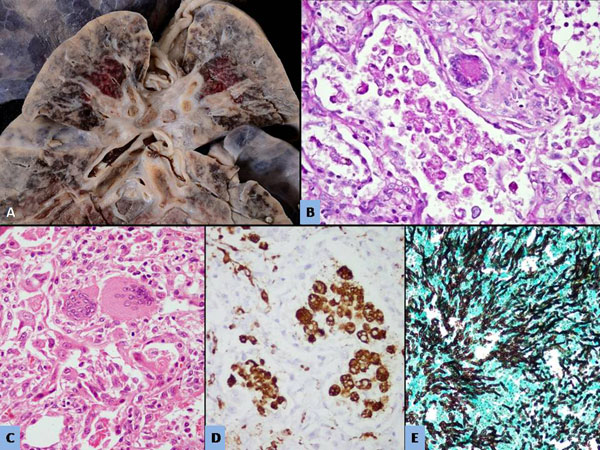 |
|
Fig. 1 (a) Cut surface of lungs with
diffuse consolidation and inspissated secretions within the
airways; (b): The alveoli demonstrating periodic acid-Schiff
(PAS) positive pigmented macrophages within its lumen (PAS x400,
original magnification); (c): Many multinucleated giant cells
with ill-formed epithelioid granulomas noted within interstitium
of the lung (H&E × 400); (d): CD 68 immunostain highlighting the
macrophages (immunoperoxidase × 400); (e): Methamine silver
stain highlighting the septate, slender Aspegillus hyphae. (Methamine
silver × 200)
|
Heart weighed 60 g; the anterior and posterior
pericardial surfaces were normal. Right ventricular dilatation was
noted. There was discoloration of the left ventricular wall along inflow
and outflow tract (Fig. 2a). On histology,
pericarditis was identified which featured similar macrophages, and few
giant cells. The inflammation was extending to underlying myocardium
which revealed myocyte loss, necrosis and interstitial edema. Many
well-formed granulomas associated with giant cells were identified in
the myocardium of both ventricles including papillary muscles and atrium
(Fig. 2b and 2c). The infiltrate was
chiefly composed of macrophages (highlighted by CD68) and few
lymphocytes (CD3+). PCR done on heart tissue for Coxsackie was negative.
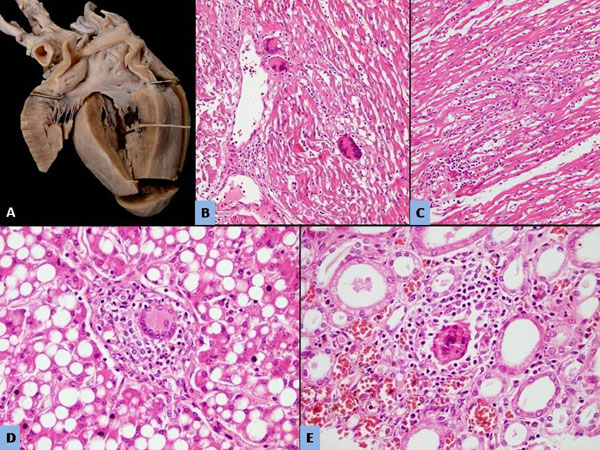 |
|
Fig. 2 (a): Gross photograph of heart
depicting discoloration of the left ventricular wall; (b and c):
Well-formed granulomas associated with giant cells identified in
the myocardium of both ventricles (H&E × 200); (d): Well-formed
granulomas with giant cells within the portal tracts (H&E ×
400); (e): Similar granulomas within the interstitium of kidneys
(H&E × 400).
|
Liver weighed 250 g with pale appearance suggestive
of fatty change. Microscopically, macrovesicular steatosis and
canalicular cholestasis was noted. Many granulomas were identified
within the portal tracts (Fig 2d), and lobule
composed of similar cellular infiltrate. Spleen weighed 21 gram was
within normal limits both on gross and microscopic examination. The
lymphoid population was preserved. Kidneys weighed 105 gms with pale and
blotchy cortical surface. Similar granulomas were identified in the
interstitium wherein the macrophages were filled with similar material (Fig
2e). Additionally glomerular immaturity (70-80% immature
glomeruli) with pigment cast nephropathy was noted with proximal tubules
in the cortex filled with pigmented casts. Occasional thrombi were
identified in one of the tributaries of renal veins. Sections from the
entire gastrointestinal tract, including stomach, esophagus, small and
large intestine were normal. No pigmented macrophages were noted within
the lamina propria. Granulomas were also identified in random sections
taken from skeletal muscle, connective tissue around cartilage, and
subcutaneous fat in section from the abdominal wall (Fig 3a-c).
Bone marrow, thymus and mesenteric and hilar lymph nodes showed
preserved T and B cell population highlighted by CD3 and CD20
immunostains (Fig 3 d-f). Hemophagocytosis was
noted in lymph nodes and spleen.
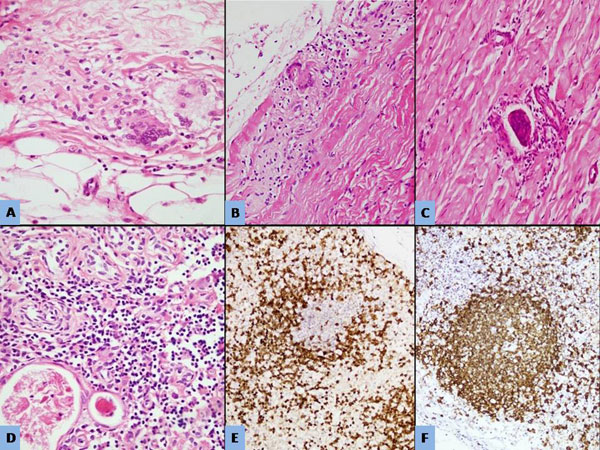 |
|
Fig. 3 Granulomas with giant cells within the
subcutaneous fat (A); Skeletal muscle(b); connective tissue (c)
(H&E x400). Thymus depicting preserved T and B cell population
(d: H&E x400);CD3 immunostain highlighting the preserved T cell
population (e: immunoperoxidase x200); CD20 immunostain
highlighting the preserved B cell (f: immunoperoxidase x200).
|
Overall autopsy diagnosis
• Granulomatous inflammation admixed with
pigmented macrophages involving lungs, heart, liver, kidneys,
Cartilage, skin and skeletal muscle — consistent with Chronic
Granulomatous disease
• Invasive Aspergillosis in lungs
• Hemophagocytosis (Lymph node and spleen)
• Ascites
Severe combined immunodeficiency (SCID) was unlikely
due to normal thymus and preserved lymphoid population within the
lymphoid organs.
Open Forum
Pediatrician 1: The fact that the child remained
well for initial 5 months of life virtually rules out usual kinds of
severe combined immunodeficiency. The deterioration started in second
half of infancy. Humoral immunodeficiencies are the commonest group to
appear at this age. Humoral immunodeficiency was ruled out by doing
immunoglobulin levels which were elevated. CGD can have two kinds of
presentation. Usual presentation is what was seen in this child, a slow,
smouldering course. It typically presents with a consolidation in the
lungs which does not resolve. Fine needle aspiration of the lung lesion
reveals fungus. Children with X-linked CGD can have very rapid,
fulminant course. This case fits with CGD, and because the child had
autosomal recessive form of CGD, the dissease course was not rapid. The
complement deficiencies present as a catastrophe with rapid downhill
course and these children usually do not survive the episodes of
infections. The pathological findings in this child were consistent with
those for CGD, but we cannot label this child as having CGD till we have
a molecular and genetic diagnosis.
Pediatrician 2: What is the mechanism for
presence of these pigmented macrophages which have been shown so
prominently in this case?
Pathology discussant: When Pphox assays and
genetic analysis was not being widely done, the presence of these
pigmented macrophages was considered to be a very important marker for
the diagnosis of CGD. The mechanisms proposed are that the pigment is
some uncharacterized pigment from the microbes to which the phagocytes
of these children are constantly exposed to. Due to inherent defect in
phagocytosis, this pigment gets collected within the cells. It is
believed to be lipofuscin or lipofuscin-like pigment which is PAS
positive.
Pathologist 1: In my opinion, the granulomas
found in the myocardium were not because of the infection but were due
to the disease (CGD) itself.
Chairman: Because CGD is a phagocytic defect,
intracellular killing of the organisms is impaired. Consequently, the
macrophages have persistence of the organisms and their particles.
Persistence of this infectious material incites granuloma formation.
Therefore, in the end it is infection only causes formation of
granulomas.
Discussion
Chronic Granulomatous disease (CGD) is an inherited
disorder of phagocytic cells [4] caused by mutations in any of the five
genes encoding the various sub-units of NADPH-oxidase system [5-8]. The
resultant defect causes an inability of phagocytes to generate the
bactericidal superoxide anions and hence an inability to contain certain
infectious pathogens. Any one of five genes encoding the structural or
regulatory subunits of phagocyte NADPH oxidase complex might be
affected, of which most common is the X-linked-gp91 phox,
other four are inherited as autosomal recessive [5-8]. The disease is
relatively uncommon, affecting 1in 2,00,000 and 1 in 2,50,000 live
births [9]. It manifests in infancy or childhood with repeated, severe
bacterial and fungal infections which are difficult to treat. Infections
by catalase positive organisms are most common, particularly
Staphylococcus aureus, Bukholderia cepacia , Serratia marcescens,
Nocardia and Aspergillus [9].
CGD is heterogenous in its manifestations, related to
the subtypes, and severity of associated macrophage defect. In majority
of patients, the superoxide production is undetectable resulting in
early manifestations. Rarely, there are low levels of respiratory burst
activity which may delay the manifestations into early adulthood [10].
Histological features in various organs are: active chronic
inflammation, with or without abscess or granuloma formation, and
presence of pigmented macrophages [4, 11-13]. Pigmented macrophages in
various organs, especially hepatic sinusoids and colonic mucosa, have
been described as a characteristic feature [11]. The pigment within the
macrophages is PAS-positive and proposed to be lipofuscin-like pigment,
the wear-and-tear pigment of the body. in the present case, granulomas
were identified in all the organs, including lung, liver, kidneys, skin,
connective tissue and heart. Involvement of the heart by the
granulomatous inflammation in the present case has not been described
commonly. CGD has been listed as one of the extremely rare causes of
granulomatous inflammation of the heart [14]. The nature of the cellular
infiltrate helps to differentiate this from viral myocarditis, where the
infiltrate is chiefly composed of lymphocytes in contrast to CGD where
pigmented macrophages are the predominant cells. Similar granulomatous
involvement of the connective tissue of the body, including the deeper
dermis and subcutaneous fat was another remarkable feature noted in the
present case.
References
1. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC,
Puck JM, Logan BR, et al. Establishing diagnostic criteria for
severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn
syndrome: The Primary Immune Deficiency Treatment Consortium experience.
J Allergy Clin Immunol. 2014;133:1092-8.
2. Buckley RH, Schiff RI, Schiff SE, Markert ML,
Williams LW, Harville TO, et al. Human severe combined
immunodeficiency: Genetic, phenotypic and functional diversity in one
hundred eight infants. J Pediatr. 1997;130:378-87.
3. Hotchkiss RS, Osmon SB, Chang KC, Wagner TH,
Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs
by both the death receptor and mitochondrial pathways. J Immunol.
2005;174:5110-8.
4. O’Shea PA. Chronic granulomatous disease of
childhood. Perspect Pediatr Pathol. 1982;7: 237-58.
5. Segal AW. Absence of both cytochrome b-245
subunits from neutrophils in X-linked chronic granulomatous disease.
Nature. 1987;326:88-91.
6. Baehner RL. Chronic granulomatous disease of
childhood: Clinical, pathological, biochemical, molecular, and genetic
aspects of the disease. Pediatr Pathol. 1990;10:143-53.
7. Umei T, Takeshige K, Minakami S. NADPH-binding
component of the superoxide-generating oxidase in unstimulated
neutrophils and the neutrophils from the patients with chronic
granulomatous disease. Biochem J. 1987;243:467-72.
8. Curnutte JT, Kuver R, Scott PJ. Activation of
neutrophil NADPH oxidase in a cell-free system. Partial purification of
components and characterization of the activation process. J Biol Chem.
1987;262:5563-9.
9. Winkelstein JA, Marino MC, Johnston Jr RB, Boyle
J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease.
Report on a national registry of 368 patients. Medicine (Baltimore).
2000;7:155-69.
10. Song E, Jaishankar GB, Saleh H, Jithpratuck W,
Sahni R, Krishnaswamy G. Chronic granulomatous disease: A review of the
infectious and inflammatory complications. Clin Mol Allergy. 2011;9:10.
11. Levine S, Smith VV, Malone M, Sebire NJ.
Histopathological features of chronic granulomatous disease (CGD) in
childhood. Histopathology. 2005;47:508-16.
12. Schappi MG, Smith VV, Goldblatt D, Lindley KJ,
Milla PJ. Colitis in chronic granulomatous disease. Arch Dis Child.
2001;84;147-51.
13. Schappi MG, Klein NJ, Lindley KJ, Rampling D,
Smith VV, Goldblatt D, et al. The nature of colitis in chronic
granulomatous disease. J Pediatr Gastroenterol Nutr. 2003;36;623-31.
14. Ferrans VJ, Rodríguez ER, McAllister HA Jr.
Granulomatous inflammation of the heart. Heart Vessels Suppl.
1985;1:262-70.
|
|
|
 |
|
