|
|
|
Indian Pediatr 2013;50: 693-695 |
 |
Distal Renal Tubular Acidosis with Hereditary
Spherocytosis
|
|
Rajiv Sinha *,
Indira Agarwal, Waleed M
Bawazir*# and Lesley J Bruce*
From Pediatric Nephrology Division, Department of
Pediatrics, Christian Medical College, Vellore; *Bristol Institute for
Transfusion Sciences, NHS Blood and Transplant, BS34 7QH, UK; #Department
of Biochemistry, University of Bristol, BS8 1TD,UK.
Correspondence to: Dr Rajiv Sinha, 37, G Bondel Road,
Kolkata 700 019, India.
Email: [email protected]
Received: November 01, 2012;
December 18, 2012;
Accepted: March 12, 2013.
|
|
Hereditary spherocytosis (HS) and
distal renal tubular acidosis (dRTA), although distinct entities, share
the same protein i.e. the anion exchanger1 (AE1) protein. Despite
this, their coexistence has been rarely reported. We hereby describe the
largest family to date with co-existence of dRTA and HS and discuss the
molecular basis for the co-inheritance of these conditions.
Keywords: Distal renal tubular acidosis, Familial,
Hemolysis, Spherocytosis,
|
Band 3, also known as anion exchange 1
(AE1), is a membrane glycoprotein that mediates chloride/
bicarbonate exchange. It is encoded by the human solute
carrier family 4 anion exchange member 1 (SLC4A1)
gene, which is expressed both at the red blood cell membrane
(eAE1) and at the basolateral membrane of alpha intercalated
cells in the distal tubules of the kidney (kAE1) [1,
2]. Mutations in SLC4A1 may cause both a renal
acidification defect manifesting as distal renal tubular
acidosis (dRTA), as well as red cell dysmorphology which
include hereditary spherocytosis, hereditary stomatocytosis
and South East Asian ovalocytosis [3]. The co-existence of
dRTA and ovalocytosis is common in certain geographical
areas; however, co-existence of dRTA with hereditary
spherocytosis has rarely been reported [4-8]. Here we report
3 siblings in a Southern Indian family with dRTA and
hereditary spherocytosis. The patients described here were
listed in a review of tropical dRTA [9], but a full
description of these unusual cases has not been reported.
Case Report
The first of the twin, offspring of
non-consanguineous parentage, presented at 2 years and 10
months of age with failure to thrive and features of
rickets. Evaluation, including frusemide challenge test,
showed normal vitamin D levels and renal functions, normal
anion gap hypokalemic metabolic acidosis, with high urine pH
(pH 6.0 following frusemide administration), hypercalciuria
and nephrocalcinosis and was conclusive of dRTA. Complete
blood count revealed compensated hemolysis (reticulocyte
count 5.4%, hemoglobin level 10.5 g/dL) with presence of
spherocytes and acanthocytes. The diagnosis of hereditary
spherocytosis was confirmed by positive osmotic fragility
test. Her twin sister was consulted shortly afterwards, with
similar complaints and clinical picture and was also found
to have dRTA (normal anion gap metabolic acidosis with urine
pH of 6.1 following frusemide challenge) and hereditary
spherocytosis (reticulocyte count 4.7%, hemoglobin 9.1 g/dL
and spherocytes and acanthocytes).
Both children responded to alkali
supplementation (2.5 to 3 mmoml/Kg of bicarbonate) with
normalisation of blood chemistry and significant improvement
of their growth. Their younger sibling, 14-months-old boy,
evaluated for poor weight gain, was found to have similar
clinical features and was also diagnosed to have dRTA (hypokalemic
metabolic acidosis, with high urine pH); and hereditary
spherocytosis. Physical growth and development improved
significantly, following alkali therapy, within weeks.
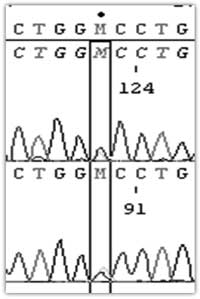
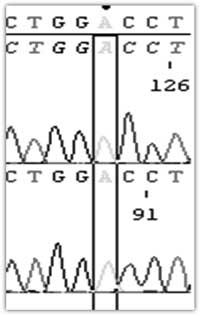
The gene encoding AE1 (SLC4A1) was
analysed (Fig. 1) and a c2573a mutation found,
resulting in the substitution of alanine by aspartic acid,
Ala858Asp. All patients were homozygous for this
substitution while their parents were heterozygous for the
same mutation. The parents had clinically normal blood
picture and renal functions.
(A) Parents
C2537
A858D
|
(B) Children
A2537
D858
 |
|
Fig. 1 DNA sequencing of
SLC4A1 shows that the parents have the heterozygous
mis-sense mutation C2537A which leads to amino acid
substitution A858D (A) whereas their children were
homozygous for the same point mutation A2537/D858
(B).
|
Discussion
AE1 is the most abundant protein on the
red cell membrane and consists of an N-terminal cytoplasmic
domain (1-399), that interacts with ankyrin in the red cell
cytoskeleton and maintains its biconcave disc shape, and a
C-terminal, membrane-spanning domain (400-911) which is
responsible for chloride-bicarbonate exchange [1]. A
truncated form of AE1, lacking the first 65 N-terminal amino
acids is present on the basolateral membrane of the
alpha-intercalated cells of the renal collecting duct where
it plays an important role in acid secretion [2,3].
Consequently mutations in SLC4A1 which codes for the
AE1 protein may have a pleiotropic effect causing both dRTA
as well as red cell dysmorphology. However the occurrence of
both effects from a single mutation is rare and was first
reported in a severe transfusion dependent patient with
spherocytosis where homozygous mutations in SLC4A1
caused complete or very severe loss of AE1 (AE1 null)
[5]. The current report discusses the largest family to date
of combined dRTA and spherocytosis secondary to a homozygous
SLC4A1 mutation. This is the second such report from
India [7] but hails from a geographically distinct area
(South India) than those previously reported (West India).
The rarity of the coexistence of both HS
and dRTA in the same patient despite sharing a common
protein has been a source of various postulations. Presence
of glycophorin A in the red cells in contrast to its absence
in the alpha – intercalated cells has been postulated as one
of the explanations. Glycophorin acts as a chaperone and
improves eAE1 trafficking to the red cell membrane.
Mutations in SLC4A1 that specifically cause dRTA
affect trafficking of kAE1 in the internal membranes, kAE1
is either held up in the endoplasmic reticulum or Golgi body
or delivered to the wrong plasma membrane, i.e. the apical
membrane instead of the basolateral membrane in
alpha-intercalated cells [10].
Apart from the AE1 null
described above, homozygous mutations, resulting in both
spherocytosis and dRTA, had been rarely reported till the
description of the homozygous mutation Ala858Asp in 2010
[7]. This was first reported in two children from different
families but from the same Maratha ethnic group in Mumbai
from Western India. Subsequently it has also been reported
from Oman [8]. Similar to these reports all the present
patients presented at an early age with features of dRTA and
showed significant reticulocytosis. The most characteristic
features of the blood film were the preponderance of
spherocytes and acanthocytes, as reported previously [7,8].
The Ala858Asp mutation is effectively a
mild mutation. A previous report showed that heterozygous
Ala858Asp individuals express 80% AE1 in their red cell
membranes, compared to normal controls, not low enough to
cause hereditary spherocytosis [4]. However homozygous
Ala858Asp patients will therefore have d" 60% AE1, a point
at which the membrane becomes unstable and hereditary
spherocytosis ensues.
Acknowledgements: Professor Oliver
Wrong initiated the collaboration that made this study
possible but passed away in February 2012 before the work
was completed. We acknowledge the contribution of Dr LK
Joseph and Dr Pragatheesh P from CMC, Vellore, India in
collecting the data for the patient and their family. The
work was supported by the UK National Health Service R & D
Directorate (LJB, WMB).
Contributors: All the authors have
contributed, designed and approved the study.
Funding: None; Competing interests:
None stated.
References
1. Schofield AE, Martin PG, Spillett D,
Tanner MJ. The structure of the human red blood cell anion
exchanger (EPB3, AE1, band 3) gene. Blood. 1994; 84:2000-12.
2. Wagner S, Vogel R, Lietzke R, Koob
R, Drenckhahn D. Immunochemical characterization of a band
3-like anion exchanger in collecting duct of human kidney.
Am J Physiol. 1987;253:F213–F21.
3. Stewart AK, Kurschat CE, Alper SL. The
SLC4 anion exchanger gene family, In: Alpern RJ,
Hebert S. (Eds), Selden and Giebisch’s "The Kidney,
Physiology and Patholophysiology" 4th. Ed Amsterdam:
Elsevier Academic Press; 2008, p.1499-1537.
4. Bruce LJ, Wrong O, Toye AM, Young MT,
Ogle G, Ismail Z, et al. Band 3 mutations, renal
tubular acidosis and South-East Asian ovalocytosis in
Malaysia and Papua New Guinea: loss of up to 95% band 3
transport in red cells. Biochem J. 2000;350:41-51.
5. Ribeiro ML, Alloisio N, Almeida H,
Gomes C, Texier P, Lemos C, et al. Severe hereditary
spherocytosis and distal renal tubular acidosis associated
with the total absence of band 3. Blood. 2000;96:1602-4.
6. Chu C, Woods N, Sawasdee N, Guizouarn
H, Pellissier B, Borgese F, et al. Band 3 Edmonton I,
a novel mutant of the anion exchanger 1 causing
spherocytosis and distal renal tubular acidosis. Biochem J.
2010;426:379-88.
7. Shmukler BE, Kedar PS, Warang P, Desai
M, Madkaikar M, Ghosh K, et al. Hemolytic anemia and
distal renal tubular acidosis in two Indian patients
homozygous for SLC4A1/AE1 mutation A858D. Am J Hematol.
2010;85:824-8.
8. Fawaz NA, Beshlawi IO, Al Zadjali S,
Al Ghaithi HK, Elnaggari MA, Elnour I, et al. dRTA
and hemolytic anemia: first detailed description of SLC4A1
A858D mutation in homozygous state. Eur J Haematol.
2012;88:350-5.
9. Khositseth S, Bruce LJ, Walsh SB,
Bawazir WM, Ogle GD, Unwin RJ, et al. Tropical distal
renal tubular acidosis: clinical and epidemiological studies
in 78 patients. QJM. 2012;105:861-77.
10. Ungsupravate D, Sawasdee N, Khositseth S,
Udomchaiprasertkul W, Khoprasert S, Li J, et al.
Impaired trafficking and intracellular retention of mutant
kidney anion exchanger 1 proteins (G701D and A858D)
associated with distal renal tubular acidosis. Mol Membr
Biol. 2010;27:92-103.
|
|
|
 |
|
