|
|
Case Reports Indian Pediatrics 2007;44:543-545 |
||
|
Congenital Perisylvian Syndrome Presenting with Intractable Seizures |
||
|
N. Hussain From the Department of Pediatric Neurology, Leicester Royal Infirmary, University Hospital of Leicester NHS Trust, Leicester LE1 5WW, United Kingdom. Correspondenee to: Dr. N. Hussain, Department of
Pediatric Neurology, Leicester Royal Infirmary, University Hospital of
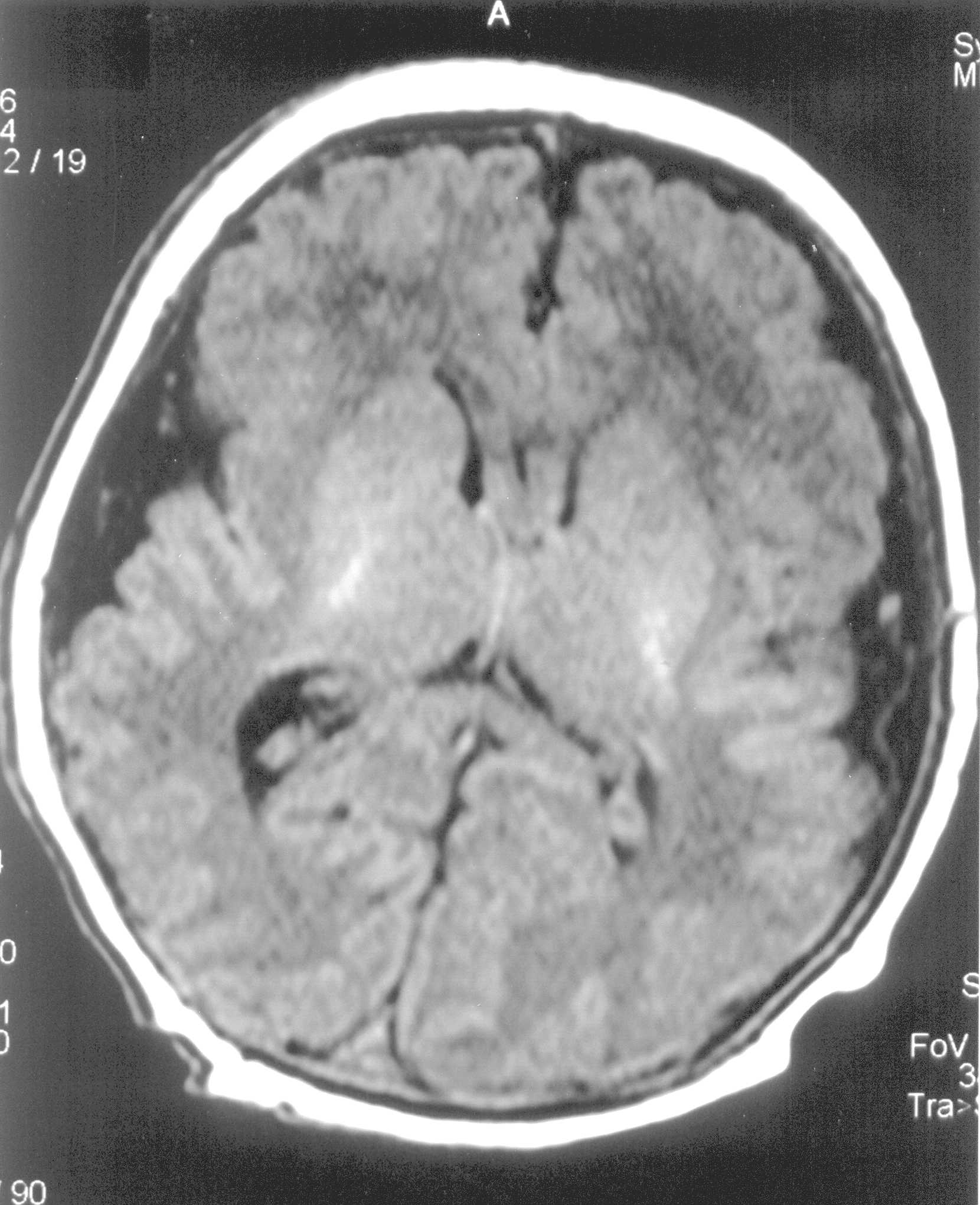
Leicester NHS Trust, Leicester LE1 5WW, Abstract: Key words: Congenital perisylvian syndrome, epilepsy, pseudobulbar. Congenital perisylvian syndrome (CBPS) is an extremely rare, late migration disorder of the brain characterized by psedobulbar palsy, mental retardation, epilepsy, and bilateral perisylvian polymicrogyria. Case Report A 9-month-old female infant presented with intractable seizures, hypotonia and feeding difficulties since birth. She was born following an unremarkable pregnancy at term by emergency caesarean section for foetal tachycardia and poor progress in labour. The blood gases of cord blood were normal. She weighed 4.2 kg (99th centile) and had an occipito-frontal head circumference of 35 cms (50th centile). She cried at birth but at around 3 minutes of age went apnoeic with stiffening of limbs. She was intubated and ventilated for 24 hrs for increasing respiratory distress and apnoeic episodes. She developed seizures on day one of life. Many types of seizures were noted, which included facial twitching, eye twitching, jerking of limbs, cycling, abnormal eye movement, eye rolling, limb stiffening, hiccupping and abnormal tonic-clonic movements. They were associated with increased heart rate and blood pressure and reduced saturation. Physical examination showed generalised hypotonia, absent gag reflexes and poor sucking. She had feeding and swallowing difficulties with protrusion and movement of the tongue moderately impaired. She required nasogastric tube feeds. Her ictal EEG was abnormal with generalised epileptiform discharges. Neurometabolic screen, which included amino acids, organic acids. lactate, ammonia, biotidinase, acylcarnitines, very long chain fatty acids, and white cell enzymes, was normal. TORCH screen was negative. Chromosome analysis was normal, 46 XX. Finally, at 4 month of age MRI brain was done, which showed thickening of the perisylvian cortex bilaterally with simplified sylvian fissures. The cortical surface was irregular and nodular. Appearances were of polymicrogyria and the distribution was consistent with bilateral perisylvian syndrome.
She has been tried on various antiepileptic drugs to control the seizures. She is currently on a poly therapy of antiepileptic medications (phenobarbitone, lamotrigine, levetiracetam) but continues to have seizures, which include absences, twitching of right side of the face and generalised tonic-clonic seizures on a daily basis. She has been started on a ketogenic diet in an attempt to control seizures. She is. now nine-months-old and has shown some improvement in her oro-motor skills. Her sucking is more consistent and prolonged. However, she still needs nasogastric tube feeds to meet her nutritive needs. She continues to be hypotonic with a head lag, has facial diplegia with paucity of voluntary facial movements, drooling of saliva and marked global developmental delay (no social smile, not started rolling over or sitting, not making any sounds). She has laryngomalacia with noisy breathing with some intercostals and subcostal recession. Her head circumference continues to grow at the 50th centile. She has been receiving physiotherapy, occupational therapy and speech and language therapist input. Discussion Congenital perisylvian syndrome (CBPS) is a migration disorder of the brain associated with distinctive clinical and imaging features. The clinical spectrum may vary from mild speech difficulties to severe disability, intractable seizures, and cognitive and behavioral problems. The exact cause is not known, although bilateral cerebral hypoperfusion, possible injury during neuronal migration, post- migational vascular accident and gene mutation are the postulated mechanisms. Relatively few pediatric cases of congenital bilateral perisylvian syndrome have been reported. Our patient had facial diplegia but no other dysmorhic features or contractures. CBPS has been associated with chromosomal abnormalities and malformations such as arthrogryposis(1), clubfeet(1), micrognathia(1), polydactyly, constriction band syndrome(2) and pituitary hypoplasia(3). Our case was hypotonic with no pyramidal signs. She had restricted tongue movements, drooling, feeding and swallowing problems and lack of speech and language development. The main differential diagnosis was cerebral palsy, Worster-Drought syndrome (WDS) and myopathy. Her electromyography (EMG), nerve conduction study NCS and muscle biopsy was normal indicating a central (cortical) cause for her problems. MRI brain helped to make an early diagnosis and differentiate it from other forms of polymicrogyria. Absence of a definite preceding perinatal insult in the clinical history and MRI changes was against a diagnosis of cerebral palsy. Worster-Drought syndrome is a spastic tetraplegic form of cerebral palsy. There is some overlap in phenotype between WDS and CBPS, which has lead some to propose that they are the same condition or possibly a continuum(4). The lack of pyramidal sign in our case makes WDS unlikely. CT scan may not always identify the polymicrogyria(5). Although it may be difficult to recognise mild forms of polymicrogyria on a magnetic resonance imaging (MRI) scan, MRI brain remain the gold standard in the diagnosis of CBPS. Increased cortical thickness with values up to 12 mm in the affected areas (normal cortical thickness varies between 1 and 4.5 mm) was identified in our case. Newer imaging techniques like the positron emission tomography (PET) scan, magneto ence-phalographic MEG scan, subtraction ictal SPECT co registered with MRI (SISCOM) have proved useful in identifying the mild forms, the epileptogenic focus, evaluating the full functional extension of the cortical anomaly and for assessing the feasibility of surgery. Linkage places the critical region for CBPS at Xq28 but the causative gene is not known at present(6). Familial, autosomal and X-linked inheritance has been reported. Genetic test in our case helped rule out the various chromosomal abnormalities associated with polymicrogyria(6). Seizures in CBPS usually begin between the ages of four to twelve years and are poorly controlled in about sixty per cent of patients. Epilepsy was found in almost 90% of cases in the series reported by Kuzniecky, et al.(1,7). Epileptic spectrum in this syndrome is broad with seizures presenting as infantile spasms, generalised tonic-clonic, typical and atypical absences, drop attacks progressing to Lennox-Gastaut syndrome(1,7). Our child presented with seizures since birth and possibly in utero (fetal tachycardia prior to birth). She had generalised epileptiform discharge on ictal EEG and multiple seizures on a daily basis, ranging from absences to generalised tonic-clonic seizures, which was resistant to medical treatment with anti epileptic drugs. Prolonged EEG and video telemetry recordings did not show non-convulsive status epilepticus. Majority of cases of CBPS have developmental, cognitive, behavioural, speech and language difficulties and epilepsy. Seizures are difficult to treat in the majority and resistant to antiepileptic medications, as in our case. Prognosis for epilepsy cannot be predicted based on the early response to treatment(8). Callosotomy has been a valuable treatment strategy in those with intractable drop attacks(1,7). Presence of esophageal malformations(9), chromosomal abnormalities and malformations may be associated with a poor prognosis. Prenatal diagnosis using fetal ultrasound or magnetic resonance imaging may be helpful but can be difficult as the regions of the brain that are involved in this malformation may not have reached their final folding until birth. In conclusion, CBPS is more common than previously thought, is recognizable by MRI brain and should be suspected clinically in any infant or child presenting with oromotor dysfunction, pseudo- bulbar signs, developmental delay and seizures.
| ||
|
References | ||
|
![]()