Juvenile idiopathic osteoporosis (JIO) is a rare
condition of unknown etiology, characterized by prepubertal onset and
spontaneous remission with progression of puberty(1). It is an important
differential diagnosis for conditions causing generalized osteoporosis
in childhood and is diagnosed by excluding other causes. In the present
communication, we report a case of JIO along with an approach to
generalized osteoporosis in childhood.
Case Report
An 8-year-old girl born to a noncon-sanguineous
married couple presented with complaints of pain in both feet on walking
and inability to get up from sitting position since the age of 3 years.
Child was apparently normal till that age, when she had an accidental
fall and sustained fracture of left femur, following which her movements
were restricted for 6 months. The examination of the child revealed
scoliosis, angulation deformity of the right arm and tenderness over
both feet. Examination of other systems was essentially normal. Routine
blood counts, urine examinations were normal. Creatinine-phosphokinase (CPK)
level was within normal limits. A radiological skeletal survey was done
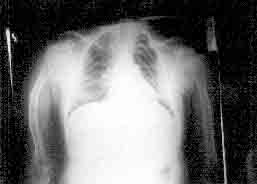
which showed pencil-thin cortices of long bones, angulation of right
humerus (Fig. 1), biconcave vertebral bodies with denser end
plates and increased intervertebral spaces. All bones had severe
osteoporosis. Serum calcium (8.1 mg/dL), phosphrous (4.3 mg/dL),
alkaline phosphatase (318 IU/L), blood urea (10 mg/dL), serum creatinine
(0.6 mg/dL), urine pH(6), and blood gases were in the normal range.
Hormonal assays showed normal pituitary, adrenal, and parathyroid
functions. Serum ceruloplasmin (17 mg/dL) and copper values (83 µg/dL)
were also normal. A diagnosis of JIO was considered.
 |
|
Fig. 1. Right humerus: Osteoporosis with pencil
thin cortex and angulation deformity. |
Discussion
Juvenile idiopathic osteoporosis is a rare form of
bone demineralization disorder. As such osteoporosis itself is a
relatively uncommon condition in childhood, and when occurs is usually
secondary to other well known causes like rickets, endocrinopathies,
malabsorption syndrome, immobilization, tumor induced, Wilson’s disease,
osteoporosis pseudoglioma syndrome, and inborn errors of metabolism like
homocystinuria. In the present case, we excluded rickets by normal
values of serum calcium, phosphorus, alkaline phosphatase, and absence
of characteristic radiological changes. Endocrinal causes were ruled out
by hormonal assays. Normal levels of serum urea, serum creatinine, blood
gases and urine pH excluded renal and metabolic causes. Wilson’s disease
was made unlikely by normal ceruloplasmin and copper values. Normal eye
examination ruled out osteoporosis pseudoglioma syndrome, another rare
condition causing generalized osteoporosis. After excluding these
conditions, we considered two primary demineralization disorders
occurring in the childhood viz osteogenesis imperfecta (OI) and juvenile
idiopathic osteoporosis. The former is a heritable condition occurring
in 4 types. Absence of blue sclera, deafness, dentigenesis imperfecta,
wormian bones in the skull excluded 0I types 1, 2, and 3. But type 4
cannot be differentiated easily. This difficulty of differentiating the
two conditions is very well recognized by earlier workers as well(1).
In our case, the features which go against type 4 OI are absence of
other involved members in the family, presence of white sclera since
birth, and absence of severe progressive deformation. Further, normal
width of long bones in the X-rays makes osteogenesis imperfecta a
distant diagnosis(2). Another aid which unfortunately was not available
and which could have helped us in resolving the issue to some extent was
the radiograph taken at the time of first fracture. If it had showed
normal bone density, then type 4 OI was more likely(3). One more
investigation which we have not done due to non availability is ratio of
a1 (III) to a1 (I) collagen in pepsin-digest of skin. An
increased ratio would strongly suggest mild osteogenesis imperfecta, but
a normal ratio would not definitely exclude it(1). Thus, with this
clinical exercise along with relevant investigations to exclude many
other conditions with osteoporosis, we arrived at a diagnosis of JIO,
which has been recognized as a diagnosis of exclusion(4). The child is
on regular follow up.
The exact pathogenesis of this disorder is not known
but available evidence points toward disturbed bone remodeling which
predominantly affects surfaces that are in contact with the marrow
cavity and results in a very low bone formation rate and decreased
cancellous bone volume(5). The age of onset of the disease varies from
one to thirteen years (mean 7 years). The disease shows no sex
predilection(6). The main presenting symp-toms include repeated long
bone fractures, pain in the back, and difficulty or inability to walk.
Particularly, the latter symptom has been stressed by many authors(7-8)
and was seen in our case as well. This fact emphasizes inclusion of
juvenile idiopathic osteoporosis in the differential diagnosis of a
child presenting with walking difficulty after ruling neuromuscular
etiologies. Typical radio-logical changes include generalized
osteoporosis, compression of vertebral bodies and metaphyses of the
lone bones. Measurement of bone mineral density will show strikingly low
values. In majority of cases, the disease remits during or after
puberty. However, exaggerated bone resorption causing vertebral
fractures during pregnancy in a recovered patient with JIO has been
observed recently(9).
Although natural remission is the rule, currently
affected children should be protected from developing permanent
deformities of the spine and long bones by restricting activities. Many
drugs like calcitriol, biphos-phonates, fluorides and calcitonin have
been used with equivocal results(4). In one study, 3 out of 4 affected
children treated with calcitriol showed significant improvement in bone
mineralization after 12 months(10). The untreated child did not show any
improvement in the same period of time. Another study on effect of
calcitonin therapy did not show any effect on the disease(11). At the
moment, experience is insufficient to advocate any treatment other than
activity restriction till natural remission.
Contributors: MLK conceived the idea and drafted
the paper. KSK worked up the case and searched the literature. Both
authors approved the final manuscript.
Competing interest: None.
Funding: None stated.
