|
|
Case Reports Indian Pediatrics 2002; 39:681-684 |
||
|
Decreased Vision as Presenting Feature in Childhood Paraganglioma |
||
|
Pradeep Venkatesh
Paragangliomas are catecholamine producing tumors from extra-adrenal chromaffin cell rests. Similar tumors arising in the adrenal medulla are known as pheochromocytomas. They have a varied clinical presentation and contribute to about 0.1% of hypertensive patients(1). These tumors may remain undetected and are found only at autopsy. Eleven per cent of patients with a catecholamine secreting tumor have a history of blurred vision(2). However, visual loss as the initial presentation is extremely rare and has not been documented earlier. We report the case of a female child who presented to us with marked bilateral loss of vision due to hypertensive retinopathy and chronic papilledema caused by a paraganglioma arising from the presacral plexus. Case Report A 6 year old female child was referred to the retina services at our centre with an initial diagnosis of viral retinitis involving both eyes. The child had been apparently normal 20 days prior to onset of a rapid loss of vision in both eyes. There were no other ocular symptoms. On ocular examination, the child had a best corrected visual acuity of counting fingers close to the face in the right eye and finger counting at 2 meters in the left eye. Projection of rays in both eyes was accurate. Eye position and ocular motility was normal. Biomicroscopic examination of the anterior segment showed no abnormalities. Pupillary reflex to light was sluggish and ill sustained in both eyes. On ophthalmoscopic evaluation of the fundus, media was clear in both eyes and the optic nerve head showed signs of established papilledema with mild hyperemia and grossly blurred margins. There were extensive superficial retinal hemorrhages, cotton wool spots and hard exudates in the peripupillary region. In both eyes an incomplete macular star was also evident. Further examination revealed a severe attenuation of the retinal arterioles and segmental dilatation of the retinal venules bilaterally. A clinical diagnosis of grade 4 hypertensive retinopathy was made. Blood pressure recorded immediately was found to be 220/120 mmHg. There was severe tachycardia, the pulse rate being 110/minute. The child was admitted. Routine urine, hemogram and chest X-ray were normal; serum urea, cretinine and electrolytes were within the normal range. Skull radiograph and CT scan of the head revealed no abnormalities. Hypertension was meanwhile controlled with nifedepine (5 mg capsules thrice daily).
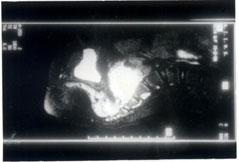
Fig 1. MRI showing a mass in relation to the presacral vertebra Later, the child’s mother informed us that the girl had intermittent mild pyrexia, along with episodes of sweating, headache and palpitations for about a year prior to the onset of visual loss.These symptoms and clinical features prompted us to consider the possibility of a catecholamine-secreting tumor as the cause of hypertension. Elevated levels of catecholamine metabolities (80 µg) were detected in the 24 hour urinary sample by biological assay (normal value less than 30 µg). This established the presence of a catecholamine secreting tumour, either an adrenal medullary pheochromocytoma or an extra-adrenal paraganglioma. Abdominal ultrasound revealed a mass in the pelvis at the presacral region. Further confirmation by magnetic resonance imaging was also undertaken (Fig. 1) The mass was excised successfully by pediatric surgeons at our institute. At surgery, the mass was reported to be about 5 cm × 5 cm in size, very vascular and lying in relation to the right iliac artery just beyond the aortic bifurcation. Histo-pathological examination of the excised mass showed features typical of a catecholamine secreting tumor. When the child came for follow up six weeks after surgery, edema of the optic nerve head was minimal and there was evolving disc pallor. Snellen visual acuity had improved to 3/60 in the right eye and 6/60 in the left eye. Discussion Pheochromocytomas are catecholamine secreting tumors arising from chromaffin cell rests situated in the adrenal medulla as well as at certain extra-adrenal sites such as the thorax, abdomen and pelvis. Tumors arising from these extra-adrenal cell rests, known as paragangliomas, account for about 10% of all pheochromocytomas. Most often, paragangliomas evolve from pelvic cell rests. In contrast to medullary pheochromocytomas, paragangliomas have a greater tendency to become malignant. Pheochromocytoma has a very varied clinical presentation. Headache is the commonest symptom and paroxysmal episodes suggestive of seizure disorder, anxiety attacks or hyperventilation are the classical manifestations(2). They may also present as sustained hypertension resistant to conventional treatment or as hypertensive crisis with encephalopathy. Rarely they manifest with symptoms of aortic dissection or myocardial infarction. Diseases like multiple endocrine neoplasia, neuro-fibromatosis and von Hippel-Lindau syndrome have also been described in association with pheochromocytoma(3). In the eye, thickened corneal nerves and conjunctival neuromas have been reported(4). Visual complications such as Horner syndrome may result as a direct consequence of the location of the tumor or due to its paraneoplastic effects(5,6). None of these associations were present in the patient reported by us. Because of their rarity and varied presentation, pheochromocytomas often remain undiagnosed and are detected only at autopsy. The diagnosis of a pheochromocytoma can be confirmed or excluded on the basis of the level of catecholamine or its metabolities in a single 24 hour urine collection, provided the patient is symptomatic or hypertensive at the time of collection of the urine sample(6,7). Surgical removal is the definitive treatment and localization of the tumor is mandatory before surgery. Removal of the tumor leads to resolution of hypertension and its ocular manifestations(8). Chen et al(9) have recently reported a case of bilateral hypertensive retinopathy secondary to pheochromocytoma in which vision returned to normal after tumor excision. In our patient however, vision improved only to 3/60 in the right eye and 6/60 in the left eye due to optic atrophy. The case reported here is unusual and has several important implications. Pheochro-mocytomas contribute to only about 0.1% cases of hypertension. Although about 11% of these patients may give a history of blurring of vision, first presentation to an ophthalmologist with a marked loss of vision has not been reported earlier. The presence of pyrexia and other symptoms like loss of weight may often lead to an initial mistaken diagnosis. If correctly diagnosed and removed surgically, pheochromocytoma has a very good prognosis, on the other hand, if the diagnosis is missed, the disease is invariably fatal. If the diagnosis is delayed and severe papilledema persists, visual loss becomes permanent owing to optic atrophy, as in this child. A high degree of suspicion and a simple measurement of blood pressure may help prevent these disastrous sequelae. Contributors: PV was involved in the clinical workup and management of patient as well as drafing of the paper. SPG and LV were involved in making the clinical diagnosis. HKT coordinated the clinical care and drafting aspects. PV will act as guarantor for the paper. Funding: None. Competing interests: None stated.
| ||
| References | ||
|
![]()