|
|
Case Reports Indian Pediatrics 2001; 38: 788-791 |
||
|
Langherhans Cell Histiocytosis in Monozygotic Twins |
||
|
From the Department
of Pediatrics, Indira Gandhi Medical College, Nagpur, India. Manuscript
received: September 25, 2000; Initial
review completed: October 31, 2000; Histiocytosis are a heterogenous group of disorders that are characterized by prolifera-tion and activation of mononuclear phagocyte system. Langerhans Cell Histiocytosis (LCH) or Class I histiocytosis is a rare disorder of unknown etiology with proliferation of Langerhan cells which may infiltrate a single or multiple organs. This disease is more common in infants and children. It is usually sporadic but a familial pattern is known. We are presenting a case of LCH in monozygotic twins which is an exceptional manifestation of this rare disease. Case Report Four-year-old monozygotic female twins were brought to our hospital with nodular swellings over the scalp and generalized lymphadenopathy since 6 weeks. They were afebrile, pale and had hepatosplenomegaly. Both had nodular lesions over the scalp and one also had supraorbital lesion (Fig. 1). There was no jaundice, petechiae, ecchymosis or joint swellings. Other systems were normal. The laboratory and radiographic investiga-tions in both children were similar. Their hemoglobin was 7 g/dl. The abdominal sono-graphy revealed hepatosplenomegaly. The skull radiograph showed multiple punched out lesions (Fig. 2). The radiography of long bones and chest were normal. The peripheral smear showed normocytic normochromic anemia with normal platelet counts and no blasts. The urine analysis, coagulation profile, bone marrow, renal and liver function tests were normal. The fine needle aspiration cytology (FNAC) and biopsy of cervical lymph node showed numerous uni, bi, and multinucleated histiocytes in the background of numerous eosinophils and lymphocytes suggestive of LCH. They were treated with Inj. Vinblastine 6 mg/m2, Tab. Prednisolone 40 mg/m2 and Tab. Endoxan 200 mg/m2 during induction phase(1). The patients are on maintenance therapy of Inj. Vinblastine 6 mg/m2 and Tab. Prednisolone. The anemia, lymphadenopathy and hepatosplenomegaly responded to treat-ment. There was shrinking of the skull lesions on radiography within 6 months of therapy.
Fig. 1. Multinodular scalp swelling in monozygotic twins.
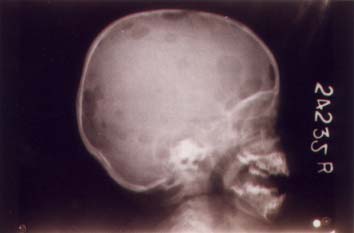
Fig. 2. X-ray skull showing multiple osteolytic shadows. Discussion Recently, histiocytosis has been classified into three classes(2). Class I and Class II are non-malignant whereas Class III is malignant. Our patients had classical manifestations of Class I or Langerhans’s cell histiocytosis in the form of multiple osteolytic lesions, lymph-adenopathy and hepatosplenomegaly without organ dysfuction. Children with organ dys-function have a higher mortality as compared to children with multiple organ involvement without dysfunction(1). LCH is a rare disease of unknown etiology. Its presentation in families particularly in twins is even less frequent. In 1952, the first concrete suggestion of genetic influence was a report on Letterer-Siwe disease in uniovular twins (3). Subsequently there have been a few reports of histiocytosis in twins(4-10). The histiocytosis was disseminated in five of these reported cases with poor response to treatment and fatality in three(6,9,10). Disseminated histio-cytosis in twins with similar clinical presenta-tion as ours has been reported earlier(4). They also had simultaneous onset of disease, almost identical clinical follow-up and findings on cranial CT. Our patients were responsive to treatment. There has been no satisfactory explanation why LCH occurs in twins. Although, it is considered to be non-hereditary, reports indicate otherwise. A survey was conducted to identify familial LCH cases (11). Nine families had more than one affected relative: five with LCH-concordant twin pairs (four were presumed monozygotic twins) and four with LCH in siblings or cousins. Another indication for genetic propensity is the occurrence of LCH in family with Hodgkins’ lymphoma. Hodgkin’s lymphoma was diagnosed in the father and the latter’s maternal uncle in one of the reported cases of LCH in twin sisters(5). Genetic susceptibility has also been suggested in Hodgkin’s disease. Monozygotic twins of patients with Hodgkin’s have been reported to have a greatly increased risk for the disease as compared to dizygotic twins. Genetic suscepti-bility also underlies Hodgkin’s disease in young adulthood(7). It is possible that a similar genetic propensity may underlie LCH. This genetic predisposition would become clearer if more cases are reported and chromosomal work-up of the families are obtained. An atypical immumological reaction has also been postulated as an underlying cause of this disease(12). Cytokines as an endogenous signal that may trigger the pathologic proliferation of Langerhans cells has also been suggested(9). Various treatment modalities have been used. The twins that were first reported in 1952 were treated with antibiotics and remained without evidence of disease 17 years later(3). Treatment modalities used nowadays are chemotherapy, radiotherapy and surgery for localized lesions. Various chemotherapeutic agents used are vinblastine, etoposide, metho-trexate, vincristine, cyclophosphamide, 6 mercatopurine, 2' deoxycoformycin, alpha-interferon, etc. (6,13,15). We used vinblastine, prednisolone and endoxan for treating LCH in these monozygotic twins who responded with-out any toxicities. A high correlation between response within 6 weeks and outcome has been reported. Our patients responded early and continue to remain well for 2 years.
Contributors: ABP worked up the case and drafted the paper. RLR
performed the literature search.
| ||
|
References | ||
|
![]()