|
Sonali
J.
Tank
Sushma Malik
Surekha M. Joshi
Medha A. Gaval
Prema Varthakavi*
From the Department of Pediatrics and *Endocrinol- ogy, T.N. Medical College and B. YL Nair Chari- table Hospital, Mumbai 400 008, India.
Reprint requests: Dr. Sonali J. Tank, 601, Sunflower, Rajawadi, Road No.2, Ghatkopar(East) Mumbai 400077, India.
Manuscript Received: September 15, 1998; Initial review completed: November II, 1998; Revision Accepted: February 22, 1999
The exact incidence of hypoparathyroidism in the pediatric population is unknown. However it is more commonly encountered in children as compared to adults, in whom primary hyperparathyroidism in seen often (1). Hypoparathyroidism should be kept in mind whenever a patient presents with late onset neonatal hypocalcemia or persistent hypocalcemia. Here we report a case of familial isolated hypoparathyroidism who presented with persistent hypocalcemia to us.
Case Report
2
A 11/2-month-male child born of a nonconsanguineous marriage was admitted with multiple episodes of generalized tonic convulsions, refusal to feed, and history of not recognizing mother and loss of social smile for few days. There was no history of fever or otorrhea. The patient was a full term normal delivery with no antenatal complications and was on exclusive breastfeeding. At the age of 3 weeks, patient was admitted in a private hospital with multifocal convulsions, was treated
as a case of meningitis with hypocalcemia and calcium supplements were given to both, mother and child. There was a history of an older sib diagnosed as hypocalcemic convulsions at 2 months of age who succumbed to pneumonia at the age of 1 year. General and systemic examination was essentially normal and there were no dysmorphic facies, cataracts, evidence of rickets or mucocutaneous candidiasis.
Biochemical investigations revealed a normal hemogram, blood glucose, serum electrolytes and a normal serum albumin (3.4 g/dl), but the serum calcium was low (6.6 mg/dl), serum phosphorus was high (6.5 mg/dl) and serum alkaline phosphates was normal (4.3 Bu). As the patient had a second episode of hypocalcemia despite calcium supplements, other etiologies for this were sought for. The serum magnesium (2.1 mg/ dl), CT scan (no basal ganglia calcification), audiometry, EEG, ECG and cardiac evaluation were all normal. Serum parathyroid hormone
revealed a low level of 1 pg/ml (normal
=
12-72 pg/ml). Thyroid function
tests, serum cortisol and renal function tests were normal. The mother's calcium, phosphorus and alkaline phosphatase, serum parathyroid hormone, thyroid function tests cortisol and blood sugar were normal. With the above history of persistent hypocalcemia, low serum parathormone, history of hypocalcemia in the older sibling and an euparathyroid mother, a presumptive diagnosis of familial isolated hypoparathyroidism was made, as we did not have the laboratory values of the sibling. Besides the initial treatment with intravenous calcium, the patient was given oral calcium (200 mg/kg elemental) and vitamin D given as calciferol (Vitamin D2) 50,000 IV/day. The serum calcium was monitored over 8-10 weeks and the patient ultimately required ] 000 mg elemental calcium with 100,000 IV vitamin D/day to maintain a serum calcium of 8.5 mg/dl. By this time the patient had
achieved social smile and was recognizing his mother. Initial follow up involved monthly visits with a monitoring of serum and urinary calcium and urine creatinine. The patient is now 10 months old and has achieved normal milestones for his age and is seizure free on calcium and vitamin D supplements.
Discussion
Hypoparathyroidism is a clinical disorder that manifests when insufficient parathyroid hormone is produced or there is decreased peipheral action of the hormone on its target organs(l-3). Either of these can result in hypocalcemia, which is primarily responsible for the production of the clinical features such as seizures, tetany, paraesthesias or muscle cramps(3,4).
Parathyroid deficient hypoparathyroidism can be nonfamilial or familial and the latter is often associated with other disorders and anomalies like DiGeorge syndrome, sensorineural deafness, dysmorphic features and autoimmune polyglandular disease type I. The non-familial or sporadic variety can be transient
or permanent, with the former presenting either as early as late onset
neonatal hypocalcemia. The permanent variety is either isolated or associated with DiGeorge syndrome( I). Pseudohypoparathyroidism is
another entity indistinguishable from
hypo-
parathyroidism clinically (except in case of Albright's Hereditary Osteodystrophy where the patients characteristically have short stature, obesity, round facies, short necks, brachydactyly and subcutaneous calcification) and has to be ruled out in a case of persistent hypocalcemia.
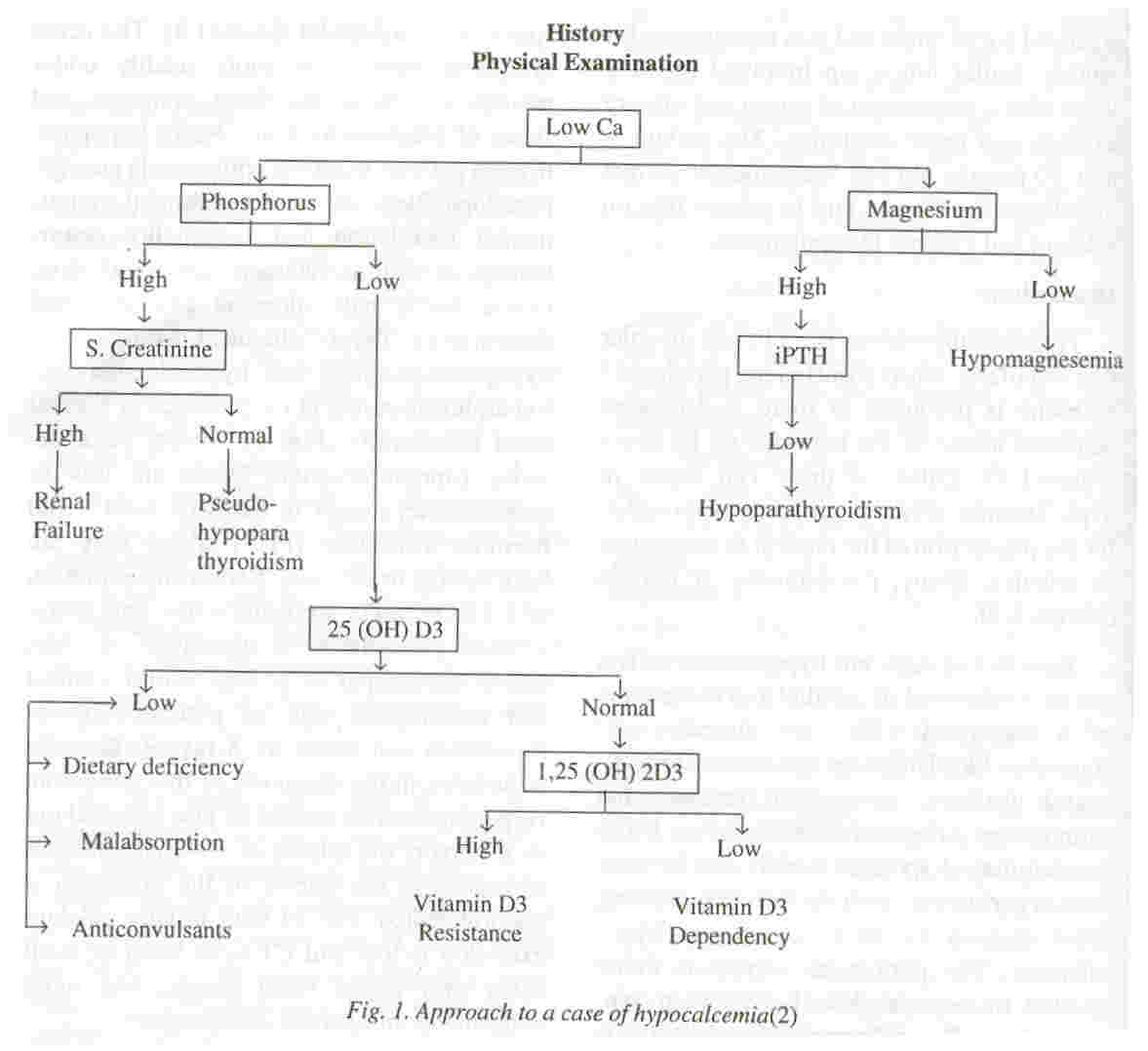
The clinical features of hypoparathyroidism of any etiology are directly attributable to hypocalcemia presenting as latent or overt tetany in the form of circumoral and digital paraesthesia, muscle cramps, laryngospasm and carpopedal spasm(l,3). The acute symptoms may occur more readily under periods of stress, increased demands and states of alkalosis(4). Chronically hypoparathyroid patients manifest with muscle cramps, pseudopapilledema, extrapyramidal signs, mental retardation and personality disturbances, as well as cataracts, dry rough skin, coarse brittle hair, alopecia and abnormal dentition(l). The biochemical hallmarks of hypoparathyroidism are hypocalcemia and hyperphosphatemia in the presence of normal renal function(3) (Fig. 1). Serum parathormone (immunoreactive) levels are low or undetectable, except in cases of parathyroid hormone resistance (PHP) where they are high normal or elevated. Serum concentration of 1,25(OH)2 D3 are usually low and serum alkaline phosphatase is normal(3). A short fourth metacarpal is a very useful clinical and radiological sign of pseudohypoparathyroidism and hence an X-ray of the wrist joint helps in the diagnosis of this condition. Hypomagnesemia should be also be ruled out as it impairs the release of PTH and induces resistance to the effects of the hormone, if uncorrected(5). The 24-hour urinary calcium excretion is low and CT scan brain or skull X-ray may detect basal ganglia and more widespread intracranial calcification(3).
The major goal of therapy in all patients irrespective of the etiology is to restore serum calcium and phosphorus as close to normal as possible. In an emergency, the acute crisis is tided over with slow intravenous calcium gluconate 1-2 ml/kg (9-18 mg/kg elemental Ca), followed by administration of vitamin D together with an oral calcium intake of 100 mg/kg elemental Calcium in divided doses(6-8).
Vitamin D2 is the commonly used vitamin D preparation as it is the least expensive and easily available but the disadvantage is its
prolonged onset of action (around 3-4 weeks) which can result in toxicity. The initial dose is 50
microgm/kg/day (2000U/kg/day) which can be gradually stepped up so as to maintain serum calcium between 8-8.5 mg/dl. 1,25- dihydroxyvitamin D3 (Rocaltrol) has become the therapy of choice for many physicians due to its shorter half life and a rapid onset of action (2-3 days) and metabolism, though it is costly. Therapy is initiated at 0.03
µg/kg/day, not exceeding a daily dose of 2
µg( 4,6,7).
Frequent monitoring of urinary and serum
calcium is recommended and periodic renal ultrasonography to detect nephrocalcinosis is advised. Additionally, calcium may be supplemeted through dairy products (milk, cheese, yoghurt) or other calcium-rich foods such as seafood (oysters, salmon, sardines), vegetables (broccoli, spinach) and nuts (apricots, dates, almonds). For patients with hyperphosphatemia, calcium rich foods as well as high phosphate foods (cola beverages) should be avoided; Use of phosphate binding antacids (aluminium hydroxide gel) may benefit. In the patients with autoimmune hypoparathyroidism, other autoimmune disorders may develop and. thus periodic reevaluation is important. In a case of suspected DiGeorge syndrome, thymic evaluation is re- quired, while patients with pseudohypoparathyroidism require evaluation of pituitary-thyroid, pituitary-gonadal and antidiuretic function(6,7).
In future, a more appropriate therapy for hypoparathyroidism may be parathormone replacement as synthetic human parathormone fragments are now available. However high cost and the need for daily parenteral administration are limiting factors for this modality.
Acknowledgement
The authors wish to thank Dr. K.D. Nihalani, Dean, T.N. Medical College, for her permission to publish this article.
|
|
1. Kruse K. Disorders of Calcium and bone metabolism. ln: Clinical Pediatric Endocrinology, 3rd edn. Eds. Brook Charles GD. London Blackwell Science Ltd, 1995; pp 735-778.
2.
Grumbach MM. The endocrine system. In: Rudolph's Pediatrics, 20th edn. Eds. Rudolph AM, Hoffman HE, Rudolph CD. London,
Prentice Hall International, 1996; pp 1841- 1844.
3.
Goltzman D, Cole DEe. Hypoparathyroidism. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 3rd edn. Ed. Favus M1. Philadelphia, Lippincott-Raven, 1996; pp 220-223.
4.
Marx S1. Hypoparathyroidism. 1n: Current Therapy in Endocrinology and Metabolism. Eds. Krieger DT, Bardin CWo Toronto, B.e. Decker Inc, 1985-1986; pp 329-333.
5.
DiGeorge AM. Hypoparathyroidism, In: Nelson's Textbook of Pediatrics. Eds. Behrman RE, Kliegman RM, Arvin AM, 15th end. Philadelhia, W.B. Saunders Co, 1996; pp 1606-1608.
6.
Rude RK. Hypocalcemia and hypoparathyroidism.ln: Current Therapy in Endocrinology and Metabolism, 6th end. Ed. Bardin CW St. Louis,
C.V. Mosby Co, 1997; pp 546-551.
7.
Root A W, Diamond FB, Mimouni FB. Parathyroid and Vitamin D. related disorders in children and adolescents. In: Pediatric Endocrinology 1st edn. Ed. Sperling MA. Philadelphia, W.B. Saunders Co, 1996; pp 480-487.
8.
Daneman D, Kooh SW, Fraser D. Hypoparthyroidism and pseudo hypoparathyrodisim in childhood. Clin Endocrinol Metab
1982;
11: 211-231.
|