|
|
|
Indian Pediatr 2020;57: 49-55 |
 |
Newborn Screening and Diagnosis of Infants
with Congenital Adrenal Hyperplasia
|
|
Pallavi Vats 1,
Aashima Dabas1,
Vandana Jain2,
Anju Seth3,
Sangeeta Yadav1,
Madhulika Kabra2,
Neerja Gupta2,
Preeti Singh3,
Rajni Sharma2,
Ravindra Kumar4,
Sunil K Polipalli1,
Prerna Batra5,
BK Thelma6 and
Seema Kapoor1
From Department of Pediatrics; 1Maulana
Azad Medical College and Lok Nayak Hospital, 2All India
Institute of Medical Sciences, 3Lady Hardinge Medical College
and Kalawati Saran Children’s Hospital, 4Hindu Rao Hospital,
5University College of Medical Sciences; and 6Department
of Genetics, University of Delhi; New Delhi, India.
Correspondence to: Dr Seema Kapoor, Director
Professor, Department of Pediatrics, Maulana Azad Medical College and
Lok Nayak Hospital, New Delhi, India.
Email: [email protected]
|
|
Congenital adrenal hyperplasia (CAH)
is an autosomal recessive endocrine disorder which can manifest after
birth with ambiguous genitalia and salt-wasting crisis. However, genital
ambiguity is not seen in male babies and may be mild in female babies,
leading to a missed diagnosis of classical CAH at birth. In this review,
we provide a standard operating protocol for routine newborn screening
for CAH in Indian settings. A standardization of first tier screening
tests with a single consistent set of cut-off values stratified by
gestational age is also suggested. The protocol also recommends a
two-tier protocol of initial immunoassay/time resolved fluoroimmunoassay
followed by liquid chromatography tandem mass spectrometry for
confirmation of screen positive babies, wherever feasible. Routine
molecular and genetic testing is not essential for establishing the
diagnosis in all screen positive babies, but has significant utility in
prenatal diagnosis and genetic counseling for future pregnancy.
Keywords: 17OHP, Cortisol, Fluoroimmunoassay,
Tandem mass spectrometry.
|
|
C
ongenital Adrenal Hyperplasia (CAH) is an
autosomal recessive disorder with an incidence ranging from 1:10,000 to
1:20,000 births [1].The screen positive rate of CAH among a cohort of
104,066 babies screened at birth in India was 1 in 5762 as per a recent
report [2].The most common defect in CAH is deficiency of enzyme
21-hydroxylase caused by mutation in CYP21A2 gene, which
comprises about 95% of all forms of CAH. Inadequate cortisol leads to an
increase of ACTH which further stimulates adrenals, resulting in
hyperplasia. A defective corticosteroid and mineralocorticoid enzymatic
pathway shunts the steroid precursors to alternate derivatives like
androgens and sex hormones.
The classical variety of CAH presents early as
genital ambiguity in newborn females (due to excess sex hormones and
their derivatives) or as adrenal crisis in both boys and girls. Adrenal
crisis is characterized by insufficient corticosteroid and aldosterone
production which causes hyponatremic dehydration and shock (salt wasting
type). Patients with adequate aldosterone production without salt
wasting who have signs of prenatal virilization are termed as simple
virilizers classified under classical CAH. A mild non-classical form
(NCCAH) of the disorder is also recognized in which presentation is in
adolescence or later.
We herein provide guidance, in the Indian context,
for diagnosis and referral of babies in early infancy with classical
forms of CAH with 21-hydroxylase deficiency.
Methods
A 2017 group of experts in the field of pediatric
endocrinology and newborn screening (Delhi Pediatric Endocrinology
Newborn Screening group- DePENS) used a semi-structured search strategy
for preparing this review. The primary database used to search
information was Medline through PubMed. The search was performed in
September 2017 and updated till January 2019. Both MeSH and keyword
based inputs were searched for articles pertaining to diagnosis and
management of classical CAH with 21-hydroxylase deficiency in childhood.
Systematic reviews, meta-analysis and randomized controlled trials were
given priority. Articles pertaining to the management of CAH in
adulthood were not included.
Newborn Screening
Incorporation of screening for 21-hydroxylase
deficiency in to all newborn screening programs is recommended, wherever
feasible.
Neonatal CAH is a disease which satisfies all the
criteria under newborn screening (NBS) checklist proposed by Wilson and
Jungner [3]. NBS can help in early diagnosis, timely treatment and
correct gender assignment of babies with classicalCAH [1,4]. Male babies
with classical CAH may go undetected in the absence of genital
ambiguity. Institution of specific steroid therapy can be life-saving in
babies with salt-losing CAH where adrenal crisis may be misdiagnosed as
sepsis. In addition, NBS can recognize simple virilizing forms in male
newborn who would otherwise present later in childhood with features of
precocious puberty. The final height of affected boys may be
significantly compromised by that time due to advanced epiphyseal
maturation [5]. However, NBS may not detect non-classical forms
consistently when performed at birth [4].
The prevalence rate of CAH has shown an increase in
the post-screening era. Sweden reported an increase in prevalence of
salt-wasting CAH from 1 in 18,600 (1969-1986) to 1 in 12,800 (from
1989-1994) after introduction of NBS [6]. The incidence of CAH reported
from Australia and Italy is variable from 1 in 15,488 or 18,105 births
(in screened population) to 1 in 18,034 or 25,462 births (in unscreened
population) [7,8].The incidence of screen positive CAH among cohort of
104,066 babies screened at birth in India was 1 in 5762 as per a recent
report. There were marked regional differences with highest from Chennai
(1:2036) to lowest from Mumbai (1:9983). The incidence of salt-wasting
CAH was higher (1 in 6934) than simple virilizing type (1 in 20,801)
[2]. Another study done on a cohort of 18,300 newborn in Andhra Pradesh
showed an incidence of 1 in 2600.The screen positives were confirmed on
recall in this study [9]. A prospective study on 11,200 newborns from
Bangalore from 2007 to 2013showed a similar incidence of 1:2800
(confirmed cases) [10].
The mortality rate in CAH varies from 0-4% in
unscreened cohort [6]. NBSwill reduce time to diagnosis, duration of
hospitalization, severity of clinical manifestations, diagnostic
uncertainty and reduce mortality in cases of salt wasting crisis. The
importance of NBS to save lives in ethnic populations with high
prevalence where timely clinical diagnosis is infrequent and CAH related
deaths occur frequently, is undoubted. Thus, incorporation of screening
for CAH should be considered as a component of NBS programme.
Standardization of first-tier screening tests with a
single consistent set of cut-off values stratified by gestational age is
recommended
It is recommended that first-tier screens for CAH
employ fluoroimmunoassay to measure 17- hydroxy progesterone (17-OHP) in
dried blood spots by heel prick methodon the same filter paper cards as
are used for other tests in NBS [1].The use of cord blood is not
recommended as the level of 17-OHP is significantly high immediately
after birth [11]. Fluoroimmunoassay has supplanted radioimmunoassay and
ELISA in most NBS programs [5,12]. It is recommended that the sample
should be obtained between 24 to 72 hours of life as 17-OHP levels are
normally high at birth and decrease rapidly in the first few postnatal
days. Though in order to decrease the false positive rate it would be
ideal to collect samples after 72 hours of birth, high birth rates
necessitate screening after 24 hours or day 2 of life. Accessible births
in the rural setting as collected by paramedical health workers maybe as
delayed till 7 days. Hence it may be practical to collect samples
between 24 hours and 7 days of life. In contrast, 17-OHP levels increase
with time in affected neonates [4]. This makes diagnostic accuracy
questionable in the first couple of days which can be an issue if
newborns are discharged early. 17-OHP levels are higher in preterm, sick
and stressed babies [4,12]. The cutoff values used should be adjusted
for these factors to reduce recall rate. The combined use of gestational
age and birth weight significantly improved predictive value in NBS for
CAH [13]. However, 17-OHP, values correlate better with the gestational
age rather than birthweight. Newborn screening programs in Switzerland
and Netherlands have adopted the gestational age cut-offs which have
improved the positive predictive value of screening [14,15]. The authors
recommend the use of the gestation specific cut-offs (whole blood units
in nmol/L) for Indian newborns as shown in Table I, which
are based on data collected from a large multicentric study from Delhi
[8].The use of birthweight-based cut-offs should be done only when
accurate gestational age assessment by first trimester ultrasonography
or record of last menstrual period is not available.
TABLE I Gestatioal Age and Birthweight-based Cut-offs for Blood Levels of 17-hydroxy
Progesterone for Newborn Screening for Congenital Adrenal Hyperplasia
|
Gestational age |
Birthweight |
Birthweight |
|
(completed wk) |
< 2500 g |
≥2500 g |
|
≤32 wk |
81
|
51
|
|
33-36 wk |
42
|
37.5
|
|
≥37 wk |
37.5
|
37.5
|
|
Birthweight |
Preterm |
Term |
|
(<37 wk) |
(≥37
wk) |
|
<1000 g |
189
|
153
|
|
1000-1499 g |
82
|
71
|
|
1500-2499 g |
42
|
37.5
|
|
≥2500 g |
37.5
|
37.5
|
|
Blood values when performed between 2-7th day of life; all
values in nmoL/L (convert nmol/L to ng/mL by multiplying by
0.66). |
Blood 17-OHP values are considered borderline between
37.5-90 nmol/L and positive beyond 90 nmol/L, as per the
fluoroimmunoassay kit-cut off values [2].The newborn screening programs
in every country adopts its own cut-off value based on their population
study. Cut-offs based on weight and gestational age are given in
Table I. A multiplication factor of 0.66 with whole blood units
(in nmol/L) may be used to obtain serum units (in ng/mL) of 17-OHP
cut-offs [2].
It is recommended that infants whose mothers received
antenatal corticosteroid treatment be retested after 2 weeks or at
discharge, whichever is later
Antenatal corticosteroids used in preterm deliveries
to facilitate fetal pulmonary maturation carry higher chances of
interfering with CAH screening results as corticosteroids can cross the
placenta and suppress the fetal hypothalamic pituitary axis. This may
reduce the blood spot 17-OHP level thus leading to false negative result
when performed at discharge or within 72 hours of birth. A reduction in
serum 17OHP to upto 30% was seen after multiple courses of steroids
[16], while inconsistent results have been obtained across otherstudies
with a single course of steroids [17]. Thus, history of all
institutional deliveries should be reviewed specially of the preterm
babies for history of antenatal steroids. It is recommended that such
infants (term or preterm) should be retested after two weeks of life,
provided the baby is monitored carefully between the two screenings for
salt losses and has easy access to health care services.For preterm
babies, the cut-off should correspond to the cut-off for the corrected
gestational age at two weeks, while for term babies the cut-off remains
the same.
A two tier protocol (initial time resolved
fluoroimmuno-assay with evaluation of positive tests by liquid chroma-tography-tandem
mass spectrometry) is recommended for confirmation of all babies tested
screen positive
The first sample for NBS should be collected on DBS
after 24 hours till 72 hours of life to be processed by an initial
fluoroimmunoassay. Babies admitted in intensive care, preterms and those
whose mothers have received antenatal steroids should also have a
mandatory repeat DBS tested after 2 weeks. In borderline and positive
cases, a second repunch sample from a different circle on the same DBS
is analyzed by fluoroimmunoassay (Repunch). In case the repunch spot
also tests positive, the baby should be recalled immediately for a
repeat venous blood sample for the second tier confirmatory testing,
which is done by liquid chromatography-tandem mass spectrometry (LC-MS)
(Fig. 1). LC/MS is a diagnostic test for confirming the
screen positives which is recommended by all the newborn screening
programs. LCMS can profile the steroids separately into cortisol, 17-OH
progesterone and 17-deoxycorticosterone and can thus differentiate the
peak obtained in fluoro-immunoassay into its different components. Thus,
it is both used as a diagnostic test and for confirming the screen
positive cases.
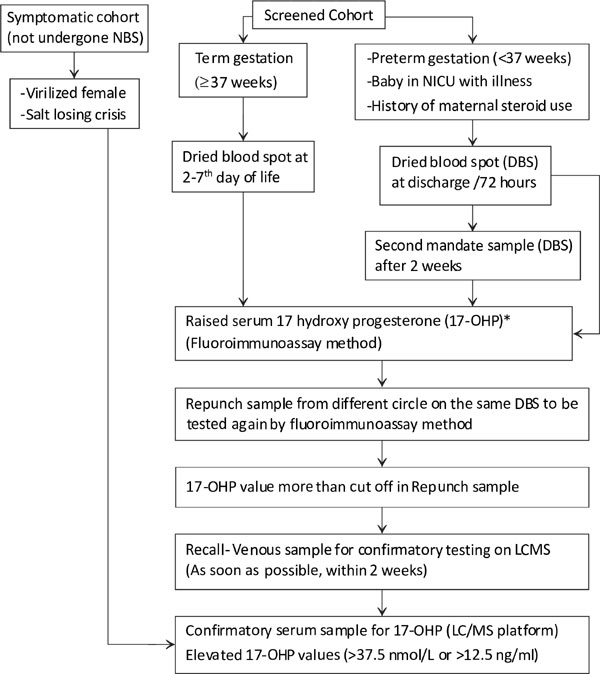
Gestational age cut offs preferred instead of birth weight
wherever available.
|
|
Fig. 1 Suggested algorithm for screening of infant
for congenital adrenal hyperplasia.
|
In order to have high sensitivity, the cut-off levels
for 17- OHP are typically set low enough to detect a positive proportion
of approximately 0.3-0.5%. A study published from the New York screening
program for CAH from 2007-14 reported a recall rate of 13,050 samples
out of 1,96,2433 samples screened (0.66%). Out of this only 105 cases
were confirmed positive (0.8% of the recalls) [18]. As the actual
prevalence of CAH is approximately 0.01 to 0.02%, this effectively means
that approximately 98% of screen positives would be false positive [19].
This in turn means that specificity of initial immunoassay is quite low
and majority of screen positive cases are false positive. The cost of
following up each false positive case could be avoided with use of a
more specific second tier test. The specific test that should be ordered
for confirmation of positive result on fluoroimmunoassay/immunoassay
should be a biochemical assay on venous sample collected after immediate
recall.
A similar two-tier protocol is being made feasible in
resource-limited settings. Positive tested samples on immunoassay can be
sent to the dedicated laboratory equipped to perform LC-MS/MS method
with a priority label from centres that do not have similar facilities.
It is recommended that LC-MS/MS be carried out on venous samples;
however, in cases where venous sample is not available, DBS samples
(whole blood) can also be used for the second tier testing.
Liquid chromatography followed by tandem mass
spectrometry (LC-MS/MS) is an alternate option where steroid ratios are
measured. This is a good confirmatory test which can be performed easily
on serum samples. The principle of organic solvent extraction increases
its specificity over an immunoassay [20,21]. Many CAH screening programs
have reported an increase in positive predictive value, from 0.8 to 7.6%
in Minnesota and 0.4 to 9.3% in Utah after implementing this approach
[22,23]. A German program tested 1609 screen positive samples(out of
242,500 samples screened) using a modified LC-MS/MS protocol which used
a ratio of the sum of 17-OHP and 21-deoxycortisol levels, divided by the
cortisol level. They concluded that a cut-off ratio of 0.53 had a
positive predictive value of 100% [24].
The downstream cost of high recall rates with
false-positive screen is difficult to estimate. The cost-effectiveness
of NBS for CAH has not been well analyzed. A false positive screen may
also be a cause of significant undue parental stress. However,
justification for NBS in CAH scores over these minor issues. Use of
better diagnostic tests will help to avert these logistic issues [5].
The use of molecular and genetic tests should be
reserved for research settings where resources permit.They are
recommended strongly for prenatal diagnosis and genetic counselling for
future pregnancies
Molecular testing can be offered to babies who test
positive on biochemical screening. The most common mutations detected
are the CYP21A2 gene mutations. One of the 10 common mutations is
present in 90% of the affected patients, thus absence of these mutations
would deem the diagnosis of CAH unlikely while presence of at least one
mutation would warrant further workup [12]. Hotspots from both the
northern and southern Indian cohort of patients with CAH have identified
a panel of 9 to 10 common mutations which can be offered to screen
positive patients in laboratories where molecular facilities are
available [2]. Genotyping of screening samples has been proposed as a
useful second tier test in lieu of LCMS in several studies [25]. Kosel,
et al. [27] in their study reported a decrease in number of
retests by 90% when the screen positive 17-OHP values were screened by
molecular means [26]. However, no large scale study has demonstrated
cost wise efficacy of this strategy. It is currently more expensive than
LC-MS/MS on a per sample basis and is not in use in any nationwide
newborn screening program.
The genotype also helps in determining the degree of
enzyme impairment, which in turn determines the severity of hormonal
abnormalities. Studies have demonstrated genotypic-phenotypic
correlation in 50% of the cases [12,27]. A deletion or intronic splice
mutation in I2G is commonly associated with salt wasting CAH, I172N
mutations and V281L were commonly associated with simple virilizing and
non-classical CAH, respectively [28]. However, genotype may not always
differentiate between salt wasting and non-salt wasting forms. For
example, patients with V281L or P30L mutations, which have been
traditionally associated with non-classical CAH may present with
virilization [29].
Thus, genotyping carries implications for prenatal
diagnosis and genetic counseling for next pregnancy.It is not required
before initiation of treatment in index case of classical CAH. It is
recommended for prenatal diagnosis or where diagnosis is questionable.
Urinary steroid profiling by Gas chromatography Mass
Spectrometry (GCMS) is not recommended as a routine confirmatory test
Urinary steroid profiling (USP) is a biochemical
analytical technique for the diagnosis of various types of
steroidogenesis defects including those leading to CAH as it can
identify and quantify a series of steroid metabolites both above and
below the enzymatic block simultaneously in a single analysis [30].
Urine steroid profiling by GCMS requires time consuming preanalytical
sample preparation. Apart from the technical challenges, an important
limitation is the inability to perform these tests in a high through put
format where large number of samples need to be processed in a short
span of time. This has limited the use of GCMS to a few experienced
research laboratories and is yet to be adapted to large scale commercial
assays [31]. It can provide a rapid simplified differential diagnosis
for CAH, where available [32].
Diagnosis in Babies Who Have Not Undergone Newborn
Screening
A morning baseline serum 17 OHP is recommended in
symptomatic individuals
A newborn who has not undergone genetic screening at
birth should be offered screening for CAH anytime he/she presents during
the neonatal period irrespective of whether symptomatic for CAH or not.
Alternately, any neonate with ambiguous genitalia or suspicion of CAH on
metabolic work-up should also be offered confirmatory testing on venous
sample for CAH on priority basis. In these babies, a single measurement
of serum 17-OHP prior to steroid administration must be sent and
interpreted.
A complete adrenocortical profile is recommended to
differentiate 21-OH deficiency from other enzyme defects and to diagnose
borderline cases. Alternatively the steroid profiling done on tandem
mass may help identify a subset of these disorders
The possibility of an alternative diagnosis other
than 21-hydroxylase deficiency may be considered in neonates/infants
with clinical or lab markers pointing to defects other than 21- OH
deficiency. The other causes of CAH include 11 b-hydroxylase
deficiency, 17 a hydroxylase deficiency, 3b hydroxysteroid dehydrogenase
deficiency and lipoid CAH. Only 21-OH deficiency and 11
b-hydroxylase
deficiency are predominantly virilizing diseases. Patients with other
causes of CAH have impaired production of cortisol by the adrenals as
well as gonadal steroids. Male patients will have undervirilization,
while female patients may or may not exhibit virilization. Clinical
features may appear similar and basal serum 17-OHP may not be fully
discriminatory in all such cases. Precursors to product ratios on
LC-MS/MS are important in differentiating the various enzyme defects. In
order to differentiate the various enzyme defects, serum 17-OHP,
cortisol, 11-deoxycortisol, 17-OH pregnenalone,
dihydroepiandrostenedione and andro-stenedione should be measured
[12,27]. The metabolites elevated in the various subtypes are shown in
Table II. Apart from 17-OH pregnenalone, the rest of tests
are available on the LC-MS/MS platform. These tests can be performed on
a venous sample collected as soon as possible but preferably within
first two weeks of life. These disorders are less common and such
children should be immediately referred to the endocrinologist for
further workup. It should be noted that thepurpose of newborn screening
is identification of babies with 21-hyroxylase deficiency, which is the
commonest.
TABLE II Subtypes of Congenital Adrenal Hyperplasia
|
Subtypes |
Phenotype |
Elevated metabolites |
|
11b-hydroxylase deficiency |
Female virilization |
Deoxycorticosterone, 11-deoxycotisol |
|
17 a-hydroxylase deficiency |
Male undervirilization |
Deoxycorticosterone, corticosterone |
|
Female virilization +/- |
|
|
3b-hydroxysteroid dehydrogenase deficiency |
Male undervirilization Female virilization +/- |
Dihydroepiandrostenedione, 17-OH pregnenalone |
Conclusions
CAH is a disease associated with significant
morbidity, mortality and long-term complications. The timely diagnosis
and treatment is challenging in the absence of newborn screening.
Screening for CAH with DBS using fluoroimmunoassays is recommended for
all babies. The confirmation of diagnosis must be made using LC-MS
method, which is getting widely available. Genetic diagnosis should be
used for diagnostic confirmation where resources permit but definitely –
for prenatal testing and counselling.
Contributors: SK,AS,VJ,MK,SY: conceived the idea;
AD,PV,PS,PB,RS,NG,RK: drafted and designed the manuscript. The group led
to the development of the manuscript and share the primary
responsibility for the final content. All authors have read and approved
the final manuscript.
Funding: None; Competing interest: None
stated.
References
1. Therrell B. Newborn screening for congenital
adrenal hyperplasia. Endocrinol Metab Clin North Am. 2001;30:15-30.
2. ICMR Task Force on Inherited Metabolic Disorders.
Newborn screening for congenital hypothyroidism and congenital adrenal
hyperplasia. Indian J Pediatr. 2018;85:935-40.
3. Wilson JMG, Jungner G. Principles and practice of
screen-ing for disease/World Health Organization, 1968. Available from:
http://apps.who.int/iris/bitstream/10665/37650/17/ WHO_PHP_34.pdf.
Accessed September 12 , 2017.
4. Pass K, Lane P, Ferhoff P, Hinton C, Panny S,
Parks J et al. US newborn screening system guidelines II:
Follow-up of children, diagnosis, management, and evaluation. Statement
of the Council of Regional Networks for Genetic Services. J Pediatr.
2000;137:S1-146.
5. Technical Report: Congenital Adrenal Hyperplasia.
American Academy of Pediatrics. Section on Endocrinology and
Committee on Genetics. Pediatrics. 2000;106:1511-21.
6. Grosse SD, Vliet VM. How many deaths can be
prevented by newborn screening for congenital adrenal hyperplasia. Horm
Res. 2007;67:284-91.
7. Gleeson HK, Wiley V, Wilcken B, Elliott E, Cowell
C, Thonsett M, et al. Two-year pilot study of newborn screening
for congenital adrenal hyperplasia in New South Wales compared with
nationwide case surveillance in Australia. J Paediatr Child
Health. 2008; 44:554-9.
8. Balsamo A, Cacciari E, Piazzi S, Cassio A, Bozza
D, Pirazzoli P, et al. Congenital adrenal hyperplasia: Neonatal
mass screening compared with clinical diagnosis only in the Emilia-
Romagna region of Italy, 1980-1995. Pediatrics. 1996;98:362-7.
9. Rama Devi AR, Naushad SM. Newborn Screening in
India. Indian J Pediatr. 2004;71:157-60.
10. Kumar RK, Das H, Kini P. Newborn screening for
congenital adrenal hyperplasia in India: What do we need to watch out
for? J Obstet Gynaecol India. 2016;66:415-9.
11. Hall K. Suitable specimen types for newborn
biochemical screening- A summary. Int J Neonatal. 2017;3:17.
12. Speiser PW, Azziz R, Laurence S, Baskin, Ghizzoni
L, Terry W. Congenital adrenal hyperplasia due to steroid 21-hydroxylase
deficiency: An Endocrine Society clinical practice guideline. J Clin
Endocr Metab. 2010;95:4133-60.
13. Olgemöller B, Roscher AA, Liebl B, Fingerhut R.
Screening for congenital adrenal hyperplasia: Adjustment of
17-hydroxyprogesterone cut-off values to both age and birth weight
markedly improves the predictive value. J Clin Endocr Metab.
2003;88:5790-4.
14. Steigert M, Schoenle E, Biason-Lauber A,
Torresani T. High reliability of neonatal screening for congenital
adrenal hyperplasia in Switzerland. J Urol. 2006;175:1118.
15. Kamp HJVD, Noordam K, Elvers B, Baarle MV, Otten
BJ, Verkerk PH. Newborn screening for congenital adrenal hyperplasia in
the Netherlands. Pediatrics. 2001;108: 1320-4.
16. Gatelais FCACA, Berthelot J, Beringue F, Descamps
P, Bonneau D, Limal J-M, et al. Effect of single and multiple
courses of prenatal corticosteroids on 17-Hydroxyprogesterone levels:
Implication for neonatal screening of congenital adrenal hyperplasia.
Pediatric Res. 2004;56:701-5.
17. Kari MA, Raivio KO, Stenman UH, Voutilinen R.
Serum cortisol, dehydroepiandrosteronesulfate, and steroid-binding
globulins in preterm neonates: Effect of gestational age and
dexamethasone therapy. Pediatr Res. 1996;40: 319-24.
18. Pearce M, DeMartini L, McMohan R, Hamel R,
Maloney B, Stansfield DM, et al. Newborn screening for congenital
adrenal hyperplasia in New York state. Mol Genet Metab Rep. 2016; 7:1-7.
19. NNSIS 2009 National Newborn Screening Information
System, NNSIS, 2009. Available from: http://www2.uthscsa.edu/nnsis.
Accessed October 14, 2018.
20. Lacey JM, Minutti CZ, Magera MJ, Tauscher AL,
Casetta B, McCann M, et al. Improved specificity of newborn
screening for congenitaladrenal hyperplasia by second-tier steroid
profiling using tandem mass spectrometry. Clin Chem. 2004;50:621-5.
21. Rauh M, Gröschl M, Rascher W, Dörr HG. Automated,
fast and sensitive quantification of 17á-hydroxy-progesterone,
androstenedione and testosterone by tandem mass spectrometry with
on-line extraction. Steroids. 2006;71:450-8.
22. Matern D, Tortorelli S, Oglesbee D, Gavrilov D,
Rinaldo P. Reduction of the false-positive rate in newborn screening by
implementation of MS/MS-based second-tier tests: The Mayo Clinic
experience (2004–2007). J Inherit Metab Dis. 2007;30:585-92.
23. Schwarz E, Liu A, Randall H, Haslip C, Keune F,
Murray M, et al. Use of steroid profiling by UPLC-MS/MS as a
second tier test in newborn screening for congenital adrenal
hyperplasia: The Utah experience. Pediatr Res. 2009;66:230-5.
24. Janzen N, Peter M, Sander S. Newbornscreening for
congenital adrenal hyperplasia: additional steroid profile using liquid
chromatography-tandem mass spectrometry. J Clin Endocrinol Metab.
2007;92:2581-9.
25. Fitness J, Dixit N, Webster D, Torresani T,
Pergolizzi R, Speiser PW, et al. Genotyping of CYP21, linked
chromosome 6p markers and a sex-specific gene in neonatal screening for
congenital adrenal hyperplasia. J Clin Endocr Metab. 1999;84:960-6.
26. Kosel S, Burggraf S, Fingerhut R, Dorr H, Roscher
A, Olgemoller B. Rapid second tier molecular genetic analysis for
congenital adrenal hyperplasia attributable to steroid 21-hydroxylase
deficiency. Clin Chemistry. 2005;51:298-304.
27. Clayton PE, Miller WL, Oberfield SE, Ritzen EM,
Sippell WG, Speiser PW. Consensus Statement on 21-Hydroxylase deficiency
from the Lawson Wilkins Pediatric Endocrine Society and the European
Society for Paediatric Endocrinology. J Clin Endocr Metab. 2002;87:
4048-53.
28. New M, Abraham M, Gonzalez B, Dumic M,
Razzaghy-Azar M, Chitayat D, et al. Genotype–phenotype
correlation in 1,507 families with congenital adrenal hyperplasia owing
to 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 2013;110:2611-16.
29. Gurgov S, Bernabé KJ, Stites J, Cunniff
CM, Lin-Su K, Felsen D, et al. Linking the degree of virilization
in females with congenital adrenal hyperplasia to genotype. Ann NY Acad
Sci. 2017;1402: 56-63.
30. Chan AO, Shek CC. Urinary steroid profiling in
the diagnosis of congenital adrenal hyperplasia and disorders of sex
development: experience of a urinary steroid referral centre in Hong
Kong. Clin Biochem. 2013;46:327-34.
31. Mc Donald J, Matthew S, Auchus R. Steroid
profiling by gas chromatography-mass spectrometry and high performance
liquid chromatography-mass spectrometry for adrenal diseases. Horm Canc.
2011;2:324-32.
32. Krone N, Hughes B, Lavery G, Stewart P, Arlt W,
Shackleton C. Gas chromatography/mass spectrometry (GC/MS) remains a
pre-eminent discovery tool in clinical steroid investigations even in
the era of fast liquid chromatography tandem mass spectrometry
(LC/MS/MS). J Steroid Biochem Mol Biol. 2010;121:496-504.
|
|
|
 |
|
