|
|
|
Indian Pediatr 2015;52: 67 -68 |
 |
Hereditary Folate Malabsorption with
Extensive Intracranial Calcification
|
|
Ikhlas Ahmad, Gousia Mukhtar, *Javed Iqbal and Syed Wajid Ali
From Departments of Pediatrics and *Neonatology, Sher-i-Kashmir
Institute of Medical Sciences, Srinagar, J&K, India.
Correspondence to: Dr Ikhlas Ahmad, Senior Resident,
Department of Pediatrics, Sher-i-Kashmir Institute of Medical Sciences,
J&K, India.
Email: [email protected]
Received: July 21, 2014;
Initial review: August 21, 2014;
Accepted: November 12, 2014.
|
|
Background: Anemia is a common
accompaniment of cerebral palsy, mental retardation and
neurodegenerative disorders. Clinical Characteristics: A
4-year-old boy with chronic megaloblastic anemia, global developmental
delay, seizures, intracranial calcification and new onset neuro-regression.
Observation: A diagnosis of hereditary folate malabsorption was
made, and he was put on oral and injectable folinic acid. Outcome:
Marked improvement at 6 month follow up. Message: Hereditary
folate malabsorption should be suspected in any child having
megaloblastic anemia and neuro degeneration disorder.
Keywords: Developmental delay, Megaloblastic
anemia, Seizures.
|
|
H
ereditary folate malabsorption (HFM) is a rare
and specific defect of the absorption of folic acid from the
gastrointestinal tract in the absence of malabsorption of any other
nutrient [1]. Findings include poor feeding, failure to thrive, and
anemia – often accompanied by leukopenia and/or thrombocytopenia – and
recurrent infections [2]. Neurologic manifestations include
developmental delay, cognitive and motor impairment, behavioral disorder
and early-onset seizures [1,3,4].
Diagnosis of HFM is confirmed by impaired absorption
of an oral folate load and low cerebrospinal fluid (CSF) folate
concentration (even after correction of the serum folate
concentration). SLC46A1, a gene encoding the proton-coupled
folate transporter (PCFT) protein, a member of the superfamily of
facilitative carriers, is associated with HFM [5-7]. We report a child
who had severe neurological involvement with relative sparing of
immunological system, and extensive intracranial calcifications.
Case Report
A 4-year-old child, product of 3rd degree
consanguineous marriage, was admitted in our hospital with suspicion of
a storage disorder in view of anemia, organomegaly and a
neurodegenerative course. He was born at term after an uneventful
pregnancy at term with no perinatal complications. At four months of
age, he was hospitalized for pneumonia and concurrent anemia. Blood
transfusion was given once. He received two more blood transfusions at
ages of 18 months and 3 years. He had repeated generalized tonic-clonic
seizures at 7 months of age when he was put on oral phenytoin. Phenytoin
was changed to sodium valproate after a diagnosis of megaloblastic
anemia was made at 18 months of age. However, seizure control was poor
despite two anticonvulsants: sodium valproate and levetiracetam at
maximum recommended doses. He also had painful oral lesions.
Developmental delay was present in all domains of development. In last
few months, he became progressively less communi-cative and showed less
interest in play; regression was most prominent in motor milestones and
was bedridden at the time of hospitalization.
 |
|
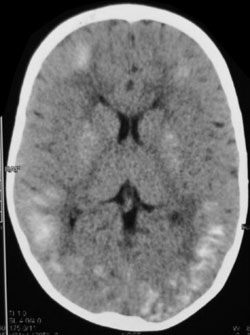
Fig. 1 Non-contrast computed
tomography of head demonstrating extensive intracranial
calcifications.
|
On examination, his weight was at the 10th centile
and his height was below the 3rd centile. He looked dull, and had severe
pallor and splenomegaly (5 cm below costal margin). His neurological
examination revealed hypotonia and hyporeflexia in all four limbs. Blood
counts revealed hemoglobin of 4.5 g/dL, total leucocyte count of 4.5 ×
10 9/L and platelet count of
87 × 109/L. Mean corpuscular
volume and mean capuscular hemoglobin were 105 and 31, respectively.
Bone marrow examination showed features of megaloblastic anemia. Serum
folate and cobalamin levels were 1 ng/mL (subnormal) and 2000 pg/mL
(above normal), respectively. Serum IgG, IgA and IgM levels were normal.
Computed tomography of head revealed diffuse intracranial calcifications
in cortex as well as in basal ganglia. Nerve conduction velocity studies
suggested peripheral neuropathy. Serum folate level after an oral
loading dose of folic acid was 3 ng/mL whereas cerebrospinal fluid (CSF)
folate level rose from 1 ng/mL to 1.5 ng/mL. Serum level was 26 ng/mL
after parenteral folinic acid. The patient was discharged on oral
folinic acid (15 mg/day) and its fortnightly injections. Parents did not
consent for repeat CSF analysis for folate measurement to guide therapy.
Six months after diagnosis, anemia, oral mucositis and splenomegaly
resolved. He had become more interactive and playful, appetite had
increased and anthropometric parameters improved. The patient had no
seizure episode over next six months. Tone in limbs improved and patient
started standing and walking with support.
Discussion
Hereditary folate malabsorption is an autosomal
recessive disorder characterized by signs and symptoms of folate
deficiency that appear within a few months after birth [7]. Infants
exhibit low blood and CSF folate levels. Treatment with folate
supplementation results in resolution of the signs and symptoms. The
disorder is caused by impaired intestinal folate absorption and impaired
transport of folate into the central nervous system [5]. Folate
deficiency results primarily in megaloblastic anemia but may affect all
three hematopoietic lineages resulting in pancytopenia [2]. Neurological
features include developmental delay, cognitive and motor impairment,
behavioral abnormalities, ataxia and other movement disorders,
peripheral neuropathy, and seizures [8-10]. Diagnosis is confirmed by
very low baseline serum folate concentrations (often <1.0 ng/mL; normal:
5-15 ng/mL) and little or no increase after an oral loading dose of
5-formyl-tetrahydrofolate. In unaffected individuals, the serum folate
concentration increases to at least 100 ng/mL [3,4,8,9]. CSF folate
concentrations are also low and may remain low after normalization of
serum folate levels.
Our patient was diagnosed when he presented with
severe neurological involvement. He had developed neurological
deterioration and was bedridden for last few months, likely due to
neuropathy. Our patient also had extensive intracranial calcifications.
Congenital infections, neurocutaneous disorders, tumors, traumatic or
ischemic insults and endocrine diseases were ruled out in our patient.
Though intracranial calcifications have been reported to occur in the
cortex or basal ganglia in hereditary folate malabsorption [2,3,9], such
extensive calcifications are unusual.
We conclude that hereditary folate malabsorption is a
treatable cause of neurological deterioration in children, and should be
suspected in any child having concomitant megaloblastic anemia.
Contributors: IA, WA: diagnosed and managed the
case; IA, GM: reviewed literature; JI: Prepared the manuscript. All
authors approved the final version of manuscript.
Funding: None; Competing interests: None
stated.
References
1. Luhby AL, Cooperman JM, Pesci-Bourel A. A new
inborn error of metabolism: Folic acid responsive megaloblastic anemia,
ataxia, mental retardation, and convulsions. J Pediatr. 1965;67:1052.
2. Jebnoun S, Kacem S, Mokrani C, Chabchoub A, Khrouf
N, Zittoun J. A family study of congenital malabsorption of folate. J
Inherit Metab Dis. 2001;24:749-50.
3. Lanzkowsky P, Erlandson ME, Bezan AI. Isolated
defect of folic acid absorption associated with mental retardation and
cerebral calcification. Blood. 1969;34:452-65.
4. Lanzkowsky P. Congenital malabsorption of folate. Am
J Med. 1970;48:580-3.
5. Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay
S, Tsai E, et al. Identification of an intestinal folate
transporter and the molecular basis for hereditary folate malabsorption. Cell.
2006;127:917-28.
6. Zhao R, Min SH, Qiu A, Sakaris A, Goldberg GL,
Sandoval C, et al. The spectrum of mutations in the PCFT gene,
coding for an intestinal folate transporter, that are the basis for
hereditary folate malabsorption. Blood. 2007; 110:1147-52.
7. Shin DS, Mahadeo K, Min SH, Diop-Bove N, Clayton
P, Zhao R, et al. Identification of novel mutations in the
proton-coupled folate transporter (PCFT-SLC46A1) associated with
hereditary folate malabsorption. Molec Genet Metab. 2011;103:33-7.
8. Geller J, Kronn D, Jayabose S, Sandoval
C. Hereditary folate malabsorption: Family report and review of the
literature. Medicine. 2002;81:51-68.
9. Corbeel L, Van den Berghe G, Jaeken J, Van Tornout
J, Eeckels R. Congenital folate malabsorption. Eur J Pediatr.
1985;143:284-90.
10. Sofer Y, Harel L, Sharkia M, Amir J, Schoenfeld
T, Straussberg R. Neurological manifestations of folate transport
defect: Case report and review of the literature. J Child Neurol.
2007;22:783-6.
|
|
|
 |
|
