|
|
|
Indian Pediatr 2015;52: 61 -62 |
 |
Association of Joubert Syndrome and
Hirschsprung Disease
|
|
Radheshyam Purkait, Rajarshi Basu, Rituparna Das and
*Uttara Chatterjee
From Department of Paediatric Medicine, NRS Medical
College and Hospital, Kolkata; and *Department of Pathology, IPGMER and
SSKM Hospital, Kolkata, India.
Correspondence to: Dr Radheshyam Purkait, Associate
Professor, Department of Paediatric Medicine, NRS Medical College &
Hospital, Kolkata 700 014, West Bengal, India.
Email:
[email protected]
Received: August 08, 2014;
Initial review: September 08, 2014;
Accepted: October 09, 2014.
|
|
Background: Association between Joubert Syndrome and Hirschsprung
disease is rare. Case characteristics: A 9-month-old girl having
developmental delay and chronic constipation. Observation: Molar
tooth sign on MRI brain and absence of ganglion cells in rectal biopsy
specimen. Outcome: Child underwent surgical repair for
Hirschsprung disease. Message: Association of these two rare
entities could be explained by ciliopathy.
Keywords: Ciliopathy, Constipation,
Developmental delay.
|
|
J
oubert syndrome is an autosomal recessive or
X-linked congenital abnormality of cerebellar vermis and brain stem that
is clinically recognized in infancy [1,2]. Hirschsprung disease is
congenital malformation of the hindgut that manifests clinically as
intestinal obstruction [3]. Association between Joubert syndrome and
Hirschsprung disease is a rare phenomenon [4]. Here we report a
9-month-old female infant who had typical clinical and radiological
findings of these two diseases.
Case Report
A 9-month-old girl was brought with the complaints of
delay in the developmental milestones, abnormal eye movements and
chronic constipation. Her parents had also noticed abnormal breathing
pattern during her early neonatal period. There was no history of
seizures and feeding or swallowing difficulty. She was born at term out
of consanguineous marriage with an uneventful perinatal period. Past
history revealed a stormy neonatal period with admission to neonatal
intensive care unit for vomiting, abdominal distension and delayed
passage of meconium. Child was diagnosed to have late-onset sepsis and
was discharged home with the advice to attend follow-up clinic at
regular interval. She had no other siblings. There was no history of
neurologic, genetic or gastrointestinal problems in other family
members.
Examination revealed facial dysmorphism in the form
of prominent forehead, high rounded eyebrows, low set ears, depressed
nasal bridge, hypertelorism, and right sided ptosis, nystagmus and
lateral squint. Abdomen was distended but there was no organomegaly.
Neurological examination showed a hypotonic child with preserved tendon
reflexes. Beside right-sided ptosis and lateral squint, examination of
the cranial nerves was normal. Rest of the systemic examination was
normal.
Complete blood count and routine biochemical tests
were within normal ranges. Thyroid profile was non-contributory.
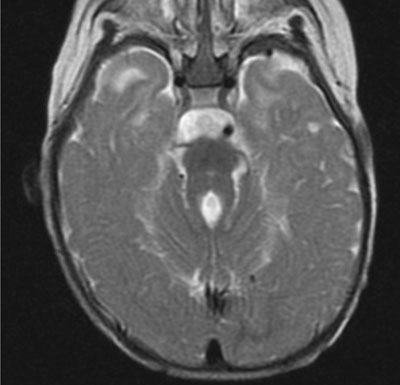
Magnetic resonance imaging (MRI) of brain revealed disorganized
cerebellar vermis with thickened superior cerebellar peduncles around
the 4th ventricle forming the classical molar tooth sign (Fig.
1). A conventional G banding chromosomal study showed a normal
karyotype (46, XX). As an initial work-up of chronic constipation, both
X-ray abdomen and ultrasonography of abdomen was done which
showed distended transverse colon, especially left lateral third along
with distended splenic flexure. Barium enema revealed distended rectum,
sigmoid colon and proximal transverse colon. Significant amount of
barium was present in the colon on twenty-four hour delayed films.
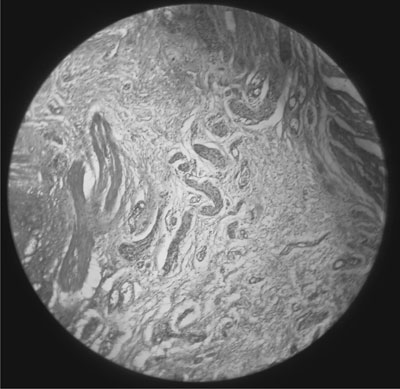
Rectal biopsy showed a large number of hypertrophied nerve bundles with
absence of ganglion cells consistent with the diagnosis of Hirschsprung
disease (Fig. 2). Subsequently child underwent definite
surgical repair for Hirschsprung disease with uneventful post-operative
period.
 |
 |
|
Fig. 1 T2W MRI showing hypoplasia of
the cerebellar vermis with elongated and thickened superior
cerebellar peduncles producing"molar tooth sign".
|
Fig. 2 Rectal suction biopsy showing a
large number of hypertrophied nerve bundles with an absence of
ganglion cells suggestive of HD [H and E stain. X10]. (See
website for color image).
|
Discussion
The neuroradiological hallmark of Joubert syndrome is
the characteristic molar tooth sign, a peculiar malformation of the
midbrain-hindbrain junction, visible on axial MRI films. Joubert
syndrome and related disorders is classified into six phenotypic
subgroups: pure, with ocular defect, with renal defect, with oculorenal
defects, with hepatic defect, and with orofaciodigital defects [2]. The
pathogenetic mechanism of Joubert syndrome includes mutation in genes
encoding proteins of the primary cilium or its apparatus. The term "ciliopathy"
usually refers to a group of genetic disorders affecting the cellular
cilia or the ciliary anchors, namely the basal bodies resulting in
improper ciliary function through proteolytic cleavage of ciliary
proteins. Proper functioning of primary cilia is known to be
instrumental in the development and functioning of several cell types,
including retinal photoreceptors, neurons, kidney tubules and bile ducts
[5,6]. These organelles have been implicated both in neuronal cell
proliferation and axonal migration [7].
We diagnosed Hirschsprung disease in our patient with
characteristic clinical, radiological and histo-pathological findings.
It is considered to be the result of premature arrest of the
craniocaudal migration of vagal neural cest cells in the hindgut between
the 5th and 12th week of gestation to form the enteric nervous system,
and is therefore regarded as a neurocristopathy [8]. Hirschsprung
disease has also been found to be associated with another ciliopathy,
Bardet-Biedl syndrome [9]. As cilia are involved in the development of
neural crest [10], ciliopathy may be the common underlying pathogenic
mechanism accountable for both the central nervous system anomalies in
Joubert syndrome and neurocristopathy in Hirschsprung disease.
Contributors: RP: diagnosed, worked up the
case and wrote the manuscript; RB, RD: managed and followed up the case;
RP, UC: reviewed the literature and prepared the final manuscript.
Funding: None; Competing interests: None
stated.
References
1. Romani M, Micalizzi A, Valente EM. Joubert
syndrome: Congenital cerebellar ataxia with the molar tooth. Lancet
Neurol. 2013;12:894-905.
2. Brancati F, Dallapiccola B, Valente EM. Joubert
Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20.
3. Amiel J, Sproat-Emison E, Garcia-Barcelo M,
Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease,
associated syndromes and genetics: A review. J Med Genet. 2008;45:1-14.
4. Ozyurek H, Kayacik OE, Gungor O, Karagoz F. Rare
association of Hirschsprung’s disease and Joubert syndrome. Eur J
Pediatr. 2008;167:475-7.
5. Lancaster MA, Gleeson JG. The primary cilium as a
cellular signaling center: Lessons from disease. Curr Opin Genet Dev.
2009;19:220-9.
6. Badano JL, Mitsuma N, Beales PL, Katsanis N. The
ciliopathies: An emerging class of human genetic disorders. Ann Rev
Genomics Hum Genet. 2006;7:125-48.
7. Millen KJ, Gleeson JG. Cerebellar development and
disease. Curr Opin Neurobiol. 2008;18:12-9.
8. Taraviras S, Pachnis V. Development of the
mammalian enteric nervous system. Curr Opin Genet Dev. 1999;9:321-7.
9. Lorda-Sanchez I, Ayuso C, Ibañez A. Situs inversus
and Hirschsprung disease: Two uncommon manifestations in Bardet-Biedl
syndrome. Am J Med Genet. 2000;90:80-1.
10. de Pontual L, Zaghloul NA, Thomas S, Davis EE,
McGaughey DM, Dollfus H, et al. Epistasis between RET and BBS
mutations modulates enteric innervation and causes syndromic
Hirschsprung disease. Proc Natl Acad Sci USA. 2009;106:13921-6.
|
|
|
 |
|
