|
|
|
Indian Pediatr 2009;46: 65-67 |
 |
Biotinidase Deficiency with Hypertonia as
Unusual Feature |
|
Narendra Rathi and Manisha Rathi
From Rathi Children and Maternity Hospital, Civil Lines,
Akola 444 001, M.S., India.
Correspondence to: Dr Narendra Rathi, Rathi Children and
Maternity Hospital, Civil Lines,
Akola 444 001, MS, India.
E mail: [email protected]
Manuscript received: March 5, 2008;
Revision accepted: March 14, 2008. |
|
Abstract
We report 3 cases of biotinidase deficiency
presenting in early infancy with neurological and cutaneous
manifestations. All of them had hypertonia (spasticity). Response to
oral biotin was excellent. One of the cases showed 7D3I biotidase
deficient mutation.
Keywords : Biotinidase deficiency, Hypertonia, Spasticity, 7D3I
mutation.
|
|
Biotinidase deficiency is one of the
treatable inherited errors of metabolism. Clinically it presents with
progressive neurological deterioration associated with cutaneous
involvement. Presence of metabolic acidosis and ketonuria substantiate the
clinical diagnosis. Final confirmation is obtained by plasma and urinary
organic acid profile and enzyme assay in cultured fibroblast. Biotin
therapy results in excellent therapeutic success with rapid normalization
of clinical and metabolic parameters. Presence of hypertonia is unusual in
these children.
Case report
Case 1: A, 3 month old boy, born out of
non-consanguineous marriage, presented with generalized tonic clonic
convulsions for one month, altered sensorium, and loss of milestones.
Convulsions were uncontrolled despite phenytoin sodium and phenobarbitone
therapy. The perinatal period was uneventful.
On examination, child was normothermic with normal
pulse and blood pressure. There was tachypnea (RR 64/min) with Kussmaul
breathing. The anterior fontanelle and fundus was normal. He had alopecia,
scanty eyebrows and blepharitis.There was seborrheic dermatitis on scalp.
He was getting seizures intermittently. He had hypertonia (spasticity) and
brisk deep tendon reflexes in all four limbs. Arterial blood gases showed
metabolic acidosis, with pH 7.14, PaCO2 12.5mm Hg, bicarbonate 4.3 mEq/L
and base deficit of –21.3. Urine examination revealed large amount of
ketones (80mg/dL). Sepsis screen, blood sugar, hemogram, serum calcium,
creatinine, sodium and potassium were normal. Cranial ultrasound was
normal. The child was treated with oxygen, parenteral fluids,
anticonvulsants and IV sodabicarb 1mL/kg. Acidosis and seizures persisted
and child lapsed into coma. A possibility of biotinidase deficiency was
kept and child was empirically put on biotin tablets through nasogastric
tube after collecting sample on filter paper. Within 24 hours, there was
dramatic response in the form of improved consciousness, decreased
acidosis and cessation of seizures. Hypertonia disappeared in 72 hours.
Anticonvulsans were withdrawn after 7 days without any recurrence of
seizures. Mass tandem spectroscopy revealed large amount of
hydroxyl-C5-acylcarnitine supporting biotinidase deficiency. The child is
on regular biotin therapy 5 mg BD and doing well. Hairs have started
growing on scalp and eyebrows.
Case 2: A 3.5 month old boy born out of
consanguineous marriage presented with seizures, regression of milestones,
altered sensorium and spasticity in upper and lower limbs. The child had
hypotrichosis on scalp and eyebrows. There was no rash or seborrhea.
Anterior fontanel, fundus and cranial ultrasound was normal. Ketonuria and
metabolic acidosis were present. Sepsis screen and serum electrolytes,
calcium, and sugar were normal. Child responded to empiric biotin therapy
5mg BD. Screening for biotinidase deficiency showed little or no enzyme
activity in the sample. DNA analysis done at Neo Gen Screening,
Bridgeville PA 15017 showed two copies (homozygous) of 7D3I Biotinidase
deficient mutation.
Case 3: A 3 months old child also presented
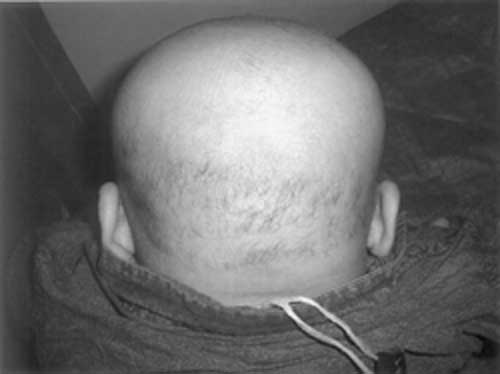
similarly with seizures, regression of milestones, alopecia (Fig.1)
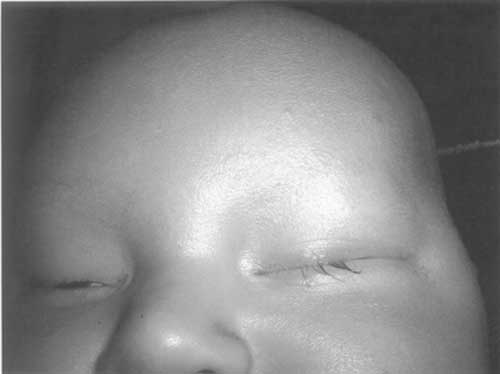
and total loss of eyebrows (Fig. 2). On examination child
had spasticity in lower limbs with normal anterior fontanel. Sepsis
screen, serum electrolytes, serum calcium, blood glucose, fundus and
cranial ultrasound were normal. Arterial blood gases and urinary ketones
were normal. Response to empiric biotin therapy was excellent. Enzyme
assay showed severe Biotinidase enzyme deficiency with enzyme activity
less than 1% of normal.
 |
 |
| Fig. 1
Alopecia. |
Fig. 2 Loss
of eyebrows. |
Discussion
Biotinidase deficiency, a recessively inherited
treatable metabolic disorder, is a defect in utilization of a water
soluble vitamin, biotin. It is an autosomal recessive disorder with
prevalence of 1 in 60000. Every 1 in 123 individuals is heterozygous for
this disorder(1). This disorder is clinically suspected in the presence of
progressive neurological deterioration (seizures, encephalopathy,
neuro-developmental delay) associated with cutaneous involvement (skin
rash, seborrhea, alopecia). The symptoms appear when the child is several
months old, which is possibly due to presence of sufficient free biotin
derived from mother.
Hypotonia is a common clinical finding in this
condition, due to reversible metabolic myopathy(2-5). Hypertonia, as
observed in these 3 cases remains unexplained. Raised intracranial
pressure was unlikely in view of normal anterior fontanel, fundus and
cranial ultrasound. In all 3 cases, hypertonia responded to Biotin therapy
in 3-5 days. Hypertonia and spasticity is described in biotinidase
deficiency only in cases which present in late childhood or
adolescence(6,7). In such cases with late presentation, spastic
paraparesis and bilateral optic atrophy are important clinical features
but hypertonia is not reported in patients who present in early infancy.
Hypertonia of extrapyramidal type (rigidity) is seen in biotin responsive
basal ganglia disease, which presents as progressive quadriparesis,
dystonia, dysarthria and subacute encephalopathy due to bilateral necrosis
of basal ganglia(8). But this disorder occurs in older children and
clinical presentation is quite different from biotinidase deficiency.
In first case, enzyme estimation could not be done.
Clinically and therapeutically, biotinidase deficiency cannot be
differentiated from holo-carboxylase synthatase deficiency, but in last 2
cases enzyme assay proved biotinidase deficiency. The most common mutation
found is deletion at chromosome 7 and insertion at chromosome 3(9), as
seen in our second case. Biotin therapy needs to be continued throughout
life. Prognosis is excellent if Biotin therapy is started early. Newborn
screening helps for prevention of neurological damage in patients with low
residual enzyme activity by early therapy. Prenatal dignosis is done by
enzyme assay in cultured amniotic cells and mutation studies. Biotin
administered prenatally is effectively taken up by the fetus and prevents
functional deficiency of carboxylases in an affected newborn(10). Genetic
counseling is offered to parents.
Biotinidase deficiency should be thought of in any
child with seizures in first few months of life associated with
encephalopathy, skin manifestations, developmental delay or regression,
metabolic acidosis and ketonuria.
Contributors : NR conceived the study and revised
the manuscript for important intellectual content. MR helped in
acquisition, analysis and interpretation of data as well as in drafting
the manuscript. NR will serve as guarantor.
Funding : None.
Competing interests : None stated.
References
1. Wolf B. Worldwide survey of neonatal screening for
biotin deficiency. J Inherit Met Dis 1991; 14: 923-927.
2. Coskun T, Tokatli A, Ozalp I. Inborn errors of
biotin metabolism. Turk J Pediatr 1994; 36: 267-278.
3. Baumgartner ER, Suormala T. MCD:Inherited and
aquired disorder of biotin metabolism. Int J Vitam Nutr Res 1997; 67:
377-384.
4. Collins JE, Nicholson NS, Dalton N, Leonard JV.
Biotinidase deficiency-early neurological presentation. Dev Med Child
Neurol 1994; 36: 268-270.
5. Bay CA, Berry GT, Glauser TA, Hayward JC, Wolf B,
Sladky JT. Reversible metabolic myopathy in biotinidase deficiency :
it’s possible role in causing hypotonia. J Inherit Metab Dis 1995;18 :
701-704.
6. Wolf B, Pomponio RJ, Norrgard KJ, Lott IT,
Baumgmartner ER, Suormala T. Delayed onset profound biotinidase
deficiency. J Pediatr 1998; 132: 362-365.
7. Ramaekers VT, Brab M, Rau G, Heimann G. Recovery
from neurological deficits following biotin treatment in a biotinidase
Km variant. Neuropediatrics 1993; 24: 98-102.
8. Ozand PT, Gascon GG, Essa MA. Biotin responsive
basal ganglia disease – A novel entity. Brain 1998; 121: 1267-1279.
9. Pomponio RJ, Reynolds TR, Cole H, Buck GA, Wolf B.
Mutational hotspot in the human biotinidase gene causes profound
biotinidase deficiency. Nature Genet 1995; 11: 96-98.
10. Suormala T, Fowler B, Jakobs C, Duran M, Lehnert W, Raab K. Late
onset HCS deficiency: pre and postnatal diagnosis and evaluation of
effectiveness of antenatal biotin therapy. Eur J Pediatr 1998; 157:
570-575. |
|
|
 |
|
