|
|
Case Reports Indian Pediatrics 2007;44:40-42 |
||
|
Glanzmann Thrombasthenia in a Neonate |
||
|
Aysegul Zenciroglu From the Department of Neonatology, and *Pediatric Hematology, Dr. Sami Ulus Children’s Hospital, Ankara, Turkey.
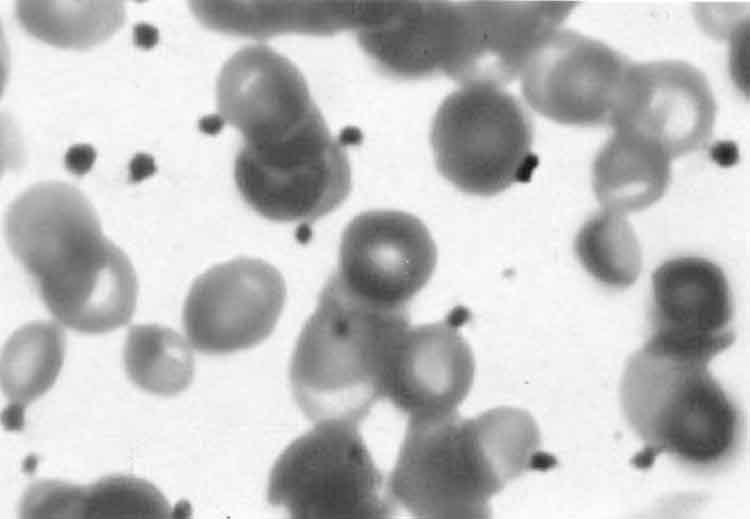
Abstract: Glanzmann thrombasthenia is a qualitative platelet function disorder manifested by skin bleeds, epistaxis, gingival bleeding, gastrointestinal hemorrhage, hematuria, hemarthrosis, intracranial hemorrhage and visceral hematomas. We report a six day old newborn presenting with hematuria following suprapubic aspiration, who was diagnosed as Glanzmann thrombasthenia. We believe it to be the youngest case reported in the literature. Key words: Glanzmann thrombasthenia, Hematuria, neonate. Glanzmann thrombasthenia is an autosomal recessive disorder associated with severe platelet dysfunction with a prolonged bleeding time and a normal platelet count. This disorder is caused by absence or deficiency of the platelet glycoprotein (GP) IIb/IIIa complex. It is associated with life long bleeding tendency, prolonged bleeding time, absent or reduced clot retraction and absent platelet aggregation with physiological agents. Clinical symptoms usually begin in infancy(1). Case Report A 6-day-old male infant was admitted to hospital with complaints of jaundice for three days. The baby was born by breech delivery after an uneventful pregnancy. He was the second child of a consanguineous marriage. The family history was unremarkable, and his older sibling was healthy. Birth weight was 3.2 kg, and Apgar scores were 9 and 8 at 1 and 5 minutes, respectively; no resuscitation effort was required. Physical examination revealed a well nourished infant with normal vital signs. Sclera and skin were icteric, examination was otherwise normal. Laboratory studies revealed total and direct bilirubin levels of 28 mg/dL and 0.8 mg/dL respectively. There was no ABO, Rh, and other blood group incompatibility. His hemoglobin level was 18.1 g/dL, hematocrit was 62%, white blood cell count was 10 × 103 /mcL, platelet count was 296 × 103 /mcL and mean platelet volume was 9.5 fl. Differential count was 52% neutrophil, 38% lymphocyte and 10% monocyte, on peripheral smear platelets were noted. Reticulocyte count was 1.2%. An exchange transfusion was performed without any complication. Postexchange serum bilirubin levels were; 13 mg/dL, 0.6 mg/dL respectively and repeat complete blood count was in the range of normal values. Blood culture yielded no microorganisms. There was no identified pathologic cause leading to hyperbilirubinemia. On day 9, abdominal ultrasonography was performed since sequestration of blood within body cavities can result increased bilirubin production. It revealed calyx echogenities of kidneys and suprapubic bladder aspiration was done to rule out a urinary tract infection. Microscopic evaluation of the urine was normal with no erythrocyte. A day after bladder aspiration, gross hematuria was noted and hemoglobin level dropped to 8.7 g/dL, and an erythrocyte transfusion was required. On peripheral blood smear, numerous platelets without clumping were noted (Fig. 1).
Prothrombin time (PT) and active partial thromboblastin time (PTT) were within normal range. Bleeding time, measured by Ivy method was 8.5 minutes (Normal values are between 1.9-5.8 minutes in newborn period)(2). The calyceal echogenities disappeared on second abdominal ultrasonography. His first peripheral blood smear was reevaluated by a pediatric hematologist and platelet aggregates were interpreted as defective. Platelet aggregation studies revealed that platelets failed to aggregate in response to ADP, epinephrine and collagen. However, ristocetin-induced platelet aggregation was normal. Flow-cytometric examination of platelets was carried out using monoclonal antibodies (Becton Dickinson. Oxford. UK) to CD41 and CD61 and results were 0.03% and 0.33%, respectively suggestive of Glanzmann thrombasthenia. Macroscopic hematuria persisted for three days while microscopic hematuria continued for five days. No microorganism was isolated from suprapubic aspiration sample. The neonate was discharged from hospital at the age of ten days. At four months old, platelet count was in normal range but peripheral blood smear revealed numerous platelets without clumping. He was followed up for two years without any bleeding problem. Discussion Glanzmann thrombasthenia is an autosomal recessive condition caused by a deficiency in platelet fibrinogen receptor glycoprotein IIb/IIIa (CD41/CD61) complex. This protein complex is a member of the integrin family of adhesion(3). Formation of complexes of GPIIb with GPIIIa protects these glycoproteins from proteolysis. A defect in either GPIIb or GPIIIa causes the degradation of the other subunit and results in the same functional defect. Most of the encountered mutations are missense mutations or deletions in the GPIIb or GPIIIa gene(4). Glanzmann thrombasthenia is classified into three subtypes. In type 1, there is a total absence of the GPIIb-IIIa complex, while in type II there is only partial deficiency of GPIIb-IIIa, usually in the range of 5-20 % of normal values. In the variant type, GPIIb-IIIa is functionally impaired(5). As CD41 was 0.03% and CD61 was 0.33%, Glanzmann thrombasthenia type 1 was diagnosed in our patient. Affected patients exhibit a lifelong moderate to severe bleeding tendency that can be diagnosed by absent platelet aggregation in response to all physiologic agonists; absent or diminished clot retraction, prolonged bleeding time, and normal platelet count(6). Epistaxis (73%), gingival bleeding (55%) and menorrhagia (98%) at menarche are the most frequent symptoms. In Glanzmann throm-basthenia, purpuric-type skin bleeding, petechiae and subconjunctival hemorrhage due to crying may be the first symptoms in infants(4). The most frequent causes of hematuria in the neonate are acute tubular necrosis after perinatal asphyxia, nephrotoxic drugs or sepsis. Other important causes of hematuria are renal vein thrombosis, urinary tract infection, trauma from catheterization or suprapubic aspiration, coagulopathy, and thrombocytopenia(7). Hematuria (6%) is not a very common symptom of Glanzmann thrombasthenia(2). In our patient, suprapubic aspiration of the bladder was easily achieved at first attempt and there was no erythrocyte on initial microscopic examination of the urine. The day after suprapubic aspiration massive hematuria requiring transfusion was noted. The most common complication following bladder aspiration is microscopic or gross hematuria, and bowel puncture. These complications are usually related to multiple unsuccessful attempts. In our case bladder aspiration was easily and safely performed and did not require multiple attempts. Red blood cells with fresh frozen plasma or fresh whole blood can be used for exchange transfusions. The former combination is deficient in platelets while the latter supplies platelets(8). Exchange transfusion was performed using fresh whole blood in our patient. There was no hemorrhage during and after exchange transfusion procedure. The diagnostic value of the bleeding time in neonates is controversial, mainly because of limited experience in executing the test. However when performed with standardized methods and techniques, the bleeding time depends only on variables such as platelets, platelet function, and vascular factors(9). On our case prolonged bleeding time, peripheral blood smear findings and low CD41/61 suggested the diagnosis of Glanzmann thrombasthenia in this patient. Contributors: AZ, AYB, ND were involved in patient management and review of literature; NY was involved in hematological workup. AYB shall act as guarantor. Funding: None. Competing interests: None stated.
| ||
|
References | ||
|
![]()