|
Personal Practice |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Indian Pediatrics 2000;37: 45-53 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Hemophilia |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Rajesh Kashyap and V.P. Choudhry From the Department of Hematology, All India Institute of Medical
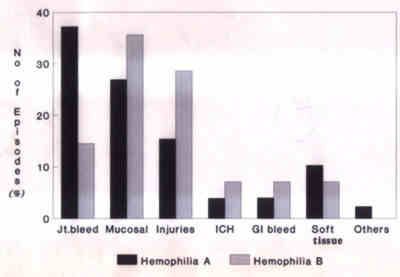
Sciences, New Delhi 110 029, India. Hemophilia A and B are x-linked bleeding disorders. The incidence of hemophilia is nearly 1 in 5000 males. It has been estimated that in India 1300 children with hemophilia are born each year and there are nearly 50,000 patients with severe hemophilia A at present(1). The greater availability and use of cryoprecipitates and factor concentrates since 1970s has greatly improved the management of hemophilia with an overall reduction in the morbidity and mortality. In the developed countries these patients have near normal life, but in the developing countries the management of these patients continues to be a major problem, as less than 5% of the affected population have adequate resources and access to the medical facilities(2,3). The clinical presentation, current concepts in the management of bleeding episodes with an emphasis on preventive measures and non-factor based therapeutic approaches are the subjects of this review. Clinical Presenation Hemophilia A or B should be suspected in any male child presenting with recurrent episodes of prolonged bleeding, occurring spontaneously or following injury or surgical procedures. A positive family history suggests the possibility of an inherited bleeding disorder. Laboratory investigations reveal isolated prolongation of activated partial thromboplastin time (APTT); the bleeding time, prothrombin time (PT) and thrombin time (TT) are normal. In hemophilia A the factor VIIIc level is low and the level of von Willebrands factor and ristocetin co-factor is normal. In hemophilia B, factor IX level is decreased. Hemophilia is classified as mild, moderate or severe based on the plasma factor levels and the severity of clinical disease correlates with the plasma factor levels. Normal plasma has clotting factor levels ranging between 50-120% (0.5 to 1.2 IU/ml) of factor activity. Patients with severe hemophilia (<1% of normal; <0.01 IU/ml) generally present with recurrent episodes of spontaneous hemarthrosis and soft tissue bleeding. Patients with moderate (1 to 5% of normal: 0.01 to 0.05 IU/ml) and mild (5 to 25% of normal; 0.05 to 0.25 IU/ml) disease have excessive bleeding following major trauma or surgery(4,5). Clinical bleeding episodes are rare during the first year of life. Joint and muscle hemorrhages are the hallmark of this disease and are often observed when the child learns to walk. Hemarthrosis develop spontaneously or following minimal trauma and the knee and elbow joints are most frequently affected, but any weight bearing joint can be affected. The joints are swollen, warm and have restricted movement. Irritability, guarding and limitation of movement of the affected joints are the initial signs of hemar-throsis in small children who cannot articulate pain. Tongue and mouth laceration is a common presentation in toddlers and occurs due to biting of tongue or lip during a fall. In societies where circumcision is customary, prolonged bleeding following the procedure may be the first presentation of this disease. Bleeding at other sites, e.g., gastrointestinal, genitourinary, retro-peritoneum are frequent in older children. Intracranial bleed (ICH) is leading cause of mortality in hemophilia patients. The incidence is 10 to 15% and the risk of ICH appears to range between 2 to 3.5% per year. Rarely, intracranial hemorrhage may occur in neonates following difficult vaginal delivery. Intraspinal hemorrhage is rare compared to ICH. The patients have acute back pain and present with paraparesis or quadriparesis depending upon the site and severity of bleed(5). The frequency of bleeding episodes at various sites in 69 hemophilic patients seen at our center between 1990 to 1994 is shown in Fig. 1.
Fig. 1. Frequency of bleeding episodes at various sites in 69 hemophilia patients followed up at AIIMS between 1990-94. Management A. General Principles It is essential to educate the patient and his family about the disease, steps to prevent the bleeding episodes and when to seek the medical care. Life of these children needs to be adjusted to minimize the risk of trauma and to prevent bleeding episodes. Periodic clinical evaluation and early institution of therapy is the key for the success of therapy. A comprehensive care of these patients involves the services of team of doctors, consisting of hematologist, physio-therapist, general surgeon, orthopedic surgeon, dentist etc.(6) The children and family members should be advised to adhere to the following precautions: 1. All intramuscular injections are contraindicated and injection should be given intravenously or subcutaneously. 2. These children should receive all recommended immunization. The injection should only be given subcutaneously using a 26 G needle and pressure applied for 2 to 3 minutes. The infants or children must be immunized against hepatitis B; recently, hepatitis A immunization has also been recommended. 3. Following precautions must be observed to prevent injuries in children: • Soft plastic or stuffed toys should be given to these children for playing. • Child should not be left unattended in an infant seat or on a raised unprotected beds. • Furniture with sharp edges must be avoided or the edges be padded. Furni-ture in the house should be kept to the minimum. • Protective elbow and knee pads can be used. • Shoes should be worn outdoors. • Children and adolescents should be encouraged to report the development of the initial subjective symptoms or objective sign of joint bleed to ensure prompt treatment. • Factor concentrate should be kept at home. Replacement therapy should be initiated at the onset of symptoms. B. Principles of Conservative Management These include the following: • These children should be treated as normal children and should have daily exercises. They should be encouraged to carry their normal physical activities and take part in sports and exercises. Sports like swimming, badminton, fishing and walking have mini-mal risk while basketball, cricket, jogging, etc. have moderate risk. The risk of injury can be reduced with proper training, pro-tective clothing and equipment. • Skin lacerations in hemophiliacs should be treated on same lines as normal persons by suturing or use of sterile adhesives. Ice wrapped in thick cloth should be applied immediately at local site to control bleeding. • A loose tooth can result in persistent blood loss and should be extracted. Pressure and ice applied to the bleeding sites and factor replacements is essential to control bleeding. • For epistaxis, the head should be slightly elevated (to 30�) and ice applied to nose. Anterior nasal packing must be done with care to control excessive bleeding. • Fluid intake should be increased in presence of hematuria to prevent clot colic. • Gauze soaked in dilute epinephrine (1:10,000 dilution) can be used for topical hemostasis to control the bleeding from mild lacerations and abrasions. Factor replace-ment may be necessary if these measures fail to control the bleeding. • Minor and recurrent joint bleeds (hemar-throsis) can be managed by keeping the joint in position of least pain along with applica-tion of ice packs, elevation and immobiliza-tion of the joint. The Don'ts of conservative treatment are: • Do not cauterize • Do not apply constricting circular bandage or wraps. • Do not scold a child for having a bleed. • Circumcision should not be done in pre-sence of family history of bleeding disorder. • Do not apply ice to skin without a protective towel or cloth. • Do not administer aspirin or related anal-gesics. C. Home Therapy The general and conservative principles of management form the basis of home care. D. Principles of Factor Replacement Therapy Patients with active bleeding whether spontaneous or following trauma and prior to surgery require factor replacement therapy. The dose of factor replacement required is deter-mined by the severity and location of bleeding and baseline activity of the different factor. The general guideline for replacement therapy in treatment of various types of bleeding episodes is given in Table I. Table I__Guideline for Factor Replacement in Management of Bleeding Episodes*
* These are only guidelines and the dose and schedule has to be modified as per severity of bleed. The duration of treatment will depend upon the individual response. ** Need for factor replacement is high initially and should be reduced gradually as indicated or depending upon the response. Dosage Calculation Factor VIII One unit of factor VIII per kilogram of body weight increases the factor VIII activity by 2%. It has a biological half life of 8 hours. The amount of factor required is calculated as: Factor VIII dose = % rise required � body weight /2 Factor IX One unit of factor IX per kilogram of body weight increases the factor IX by 1% and its half life is 16-18 hours. Factor IX dose = % rise required � body weight 1. Hemarthrosis Joints are the most common site of bleeding in hemophilic patients. The limb should be immobilized in functional position to relieve the pain and analgesics like paracetamol, para-cetamol/dextropropoxyphene can be used. The factor level should be raised to 25-35% for the first 48 hours and reduced to 15-20% for 10-15 days. Physiotherapy program must be initiated as soon as the pain subsides to strengthen the muscles, stabilize the joint and to prevent contractures. In patients who develop chronic synovitis with effusion, temporary immobiliza-tion with a removable alkathene or POP splint is useful. Contractures of the joint can be corrected by using serial wedge casts. 2. Intracranial Bleeds Intracranial bleed may be extradural, subdural or intracerebral, and may occur following trauma or spontaneously. Trauma is usually trivial and may precede the ICH by several days. Intracranial bleed must be suspected in all hemophilia patients in the following situations: (i) Presenting with altered sensorium and neurological deficit; (ii) History of headache or vomiting with or without history of trauma; and (iii) All patients with history of head trauma with or without clinical signs or symptoms of raised intracranial pressure. A high index of suspicion is the key for early diagnosis. Computerized tomography is essen-tial to confirm the diagnosis and for studying the site and size of the bleed. All patients should be given factor replacement (100% correction) as bolus dose immediately on presentation to the emergency department. Even in the absence of evidence of any bleed it is preferable to give prophylactic factor replacement (upto 50% correction) for 2 or 3 days and the patient should be sent home if there is no neurological deterioration. In patients with definitive ICH the minimum duration of treatment should be for 14-21 days. Repeat CT scan should be perform-ed to study the resolution of bleed and for deciding further treatment. Surgical treatment is only recommended if progressive neuro-logical deficit occurs inspite of medical treat-ment. In view of the high recurrence rate of ICH it is recommended that prophylactic factor therapy should be given for a period of at least 6 months. Children with recurrent episodes of ICH should ideally receive prophylactic therapy life long. All these children should be given anticonvulsants (e.g., phenytoin) for 6 months in the absence of convulsive episodes and for longer periods in the presence of convulsions. Many of these patients have severe residual neurological deficits such as ataxia, hemiparesis and aphasia and should be given adequate physical therapy and rehabilitation faci-lities(7,8). 3. Surgical Procedures Hemophilic patients may occasionally require elective surgery or rarely an emergency surgery. Any major surgery requires an active collaboration between the surgeon, hematologist and coagulation laboratory and must be performed at centers with all facilities. Therefore, the following points must be observed before undertaking surgery: (a) reassess the factor levels, (b) do screening test for presence of factor inhibitors, (c) avoid anticplatelet agents before and after surgery, (d) surgery should be scheduled early in the week and early in the day to ensure the best access to laboratory services and consultants, and (e) ensure that adequate amount of factor replacement products are available. The guide-lines for factor replacement for major surgery are given in Table II. Table II__Guideline for Factor Replacement in Hemophilic Patients Undergoing Major Surgery
4. Mucosal Bleeds Hemophilic children often present with mucosal bleeds or with epistaxsis. Antifibrino-lytic agents e-aminocaproic acid (Amicar, EACA) can be tried initially to control the bleeding for the initial 48-72 hours. The recommended dose is 50-100 mg/kg every 6 hourly as slow intravenous infusion. If this therapy fails replacement therapy can be used and the factors levels should be raised between 15-20% for 4-5 days. 5. Dental Procedures Good oral hygiene must be ensured in all patients to prevent the need for teeth extractions. Scaling and polishing of teeth usually does not require factor replacement. If dental extraction has to be done, antifibrinolytic agents should be given along with factor replacement therapy. Antifibrinolytic agent should be administered for 7-10 days, beginning the night before the procedure. The factor replacement is given as recommended in Table I. Use of fibrin glue to pack the socket after extraction has greatly helped to decrease the need for factor replacement therapy. Vicryl sutures which dissolve slowly over a three week period should preferably be used for better hemostasis. Tranexamic acid (7.8%) mouth wash along with oral tranexamic acid enhances its effect. A course of metronidazole is recommended after dental extraction(9). 6. Prophylaxis Joint disease is the major cause of morbidity of hemophilia and 90% of severe hemophilics develop arthropathy. Patients with moderate hemophilia have lower incidence of joint involvement. The aim of hemophilia prophy-laxis is to administer factor VIII or IX at frequent intervals to keep the levels above 1% which is sufficient to prevent spontaneous joint bleeds and preserve joint function. The recom-mended schedule is 25 to 40 U/kg of F VIII three times a week (hemophilia A) and 25 to 40 U/kg of F IX twice a week (hemophilia B). Ideally prophylaxis should be started by one year of age and continued indefinitely(10). Sources of Factor VIII and IX 1. Fresh Frozen Plasma (FFP) Fresh frozen plasma contains all the clotting factors in a concentration of approximately 1 unit of clotting factor activity per milliliter. Each bag of FFP contains 180-220 ml of plasma (about 180-220 units of FVIII and FIX). The large volume of FFP required to achieve the high factor levels is its main limitation. However, 10 to 15 ml/kg of FFP may be given safely in one dose. Diuretics must be given along with FFP to prevent volume overload. However, FFP still remains the chief source of factor VIII and IX in our country(2). 2. Cryoprecipitates Cryoprecipitate is prepared from FFP by slow thaw technique and is stored at 20�C. It is rich in fibrinogen, von Willebrand factor and factor VIII. It does not contain factor IX. A single unit of cryoprecipitate prepared from one unit of FFP contains 80-100 units of factor VIII. The problem with use of cryoprecipitate is: (i) it requires storage at _20�C in deep freezer; (ii) factor VIII content is not uniform in all the bags and hence it is difficult to achieve the desired factor VIII levels; and (iii) the inherent risk of blood transmitted viral and other infections continues to pose a challenge. 3. Factor Concentrates Table III__Currently Available Factor VIII and IX Concentrate Products*
* Factor VIII concentrate can be obtained from Hemophilia Federation of India or its branches. At present a variety of factor VIII and factor IX concentrates are available (Table III). Intermediate purity F VIII concentrates are prepared from cryoprecipitate, from which some of the fibrinogen is removed by a variety of methods and the specific activity of the product varies between 1-50 U/mg. High purity con-centrates are prepared directly from plasma by chromatographic separation and/or mono-clonal antibodies and the specific activity varies between 50-200 U/mg. Recently, recombinant FVIII has been made available and it has the same in vivo recovery and biological half life as that of plasma derived F VIII concentrates and have no risk of blood transmitted infections. Prothrombin complex concentrates (PCC) containing FIX has been the main source of FIX for the last three decades. However, recently high purity FIX has been made commercially available and these are prepared by use of monoclonal antibodies (Mononine) or ion-chromatography (Alpha-nine)(11). Safety of Factor Concentrates The risk of transmission of blood borne viruses is a major concern with the use of factor replacement products. The first step in elimination of this risk is to select healthy donors who are seronegative for HIV, hepatitis B and C. However, serological tests have limitations and may not detect the donors who have been recently infected. Virucidal methods have been adopted to improve the safety of factor concentrates and these are: (i) terminal heating of the lyophilized products at 80�C (dry heating); (ii) heating in solution at 60�C (pasteurization) in the presence of stabilizers or in moisture with hot vapor under high pressure; and (iii) solvent/detergent method using organic solvent (TNBP) and a detergent (sodium cholate, tween 80 or triton x-100). The risk of transmission of HIV, hepatitis B and C has decreased markedly after the adoption of these virucidal methods. However, these methods are not very effective against thermoresistant, non enveloped virus such as hepatitis A and parvovirus B 19(11). Inhibitors of Factor VIII and IX The most challenging complication of hemophilia treatment is the development of inhibitors following prolonged use of factors. Approximately 15 to 35% of patients with hemophilia A develop inhibitor antibodies. In hemophilia B patients the prevalence of inhibi-tors is low (approximately 6%). Development of inhibitor is clinically suspected when the patient does not show the expected response following factor replacement. In the laboratory, the presence of inhibitor is revealed by prolonged APTT which is not corrected by addition of normal plasma in 1:1 volume. The quantitation of an inhibitor is done by Bethesda assay. One Bethesda unit (BU) is defined as the amount of antibody needed to destroy 50% of the factor VIII/IX activity in the normal pooled plasma. Patients of hemophilia A with low inhibitor level (<5BU) can be adequately treated with increased dose of factor VIII (or factor IX in hemophilia B patients). In patients with high inhibitors (>10BU) the main stay of treatment is the bypass therapy with prothrombin complex concentrate (PCC) or activated prothrombin complex concentrate (APCC) which contain activated factor II, VII, IX and X. The usual dose is 50-100 units/kg every 6 to 8 hourly. The main disadvantage of these agents is the inability to measure its clinical efficacy by laboratory tests and an increased risk of throm-boembolic phenomenon. Recently, recombinant factor VIIa has become available, which can be effectively used for management of these cases. In patients with high F VIII inhibitors (>10BU) who do not show clinical or laboratory improvement (correction of APCC and PCC) can be treated with Porcine F VIII as it has little cross reactivity with human F VIII inhibitors. In patients who fail to respond to all these treatments, intensive plasmapheresis to remove the IgG inhibitor antibody can be performed. Immunosuppressive agents such as prednisolone, cyclosphospha-mide, azathioprine, etc. have been used to suppress the inhibitor production and produce long term remissions(12). Non Factor Based Therapeutic Agents Desmopressin Desmopressin (DDAVP, stimate) is a synthetic vasopressin analogue. It temporarily increases the plasma levels of factor VIII: c and von Willebrands factor. DDAVP when administered intravenously in the dose of 0.3 mg/kg (upto 20 mg in blood) produces 3 to 5 folds rise in the plasma levels of F VIII and von Willebrands factor. The peak factor levels are achieved at 30 to 60 minutes with a half life similar to exogenous clotting factors. It is effective in patients with mild and moderate hemophilia undergoing minor surgery or dental procedures and in management of von Willebrands disease. The side effects are minimal which include facial flushing, head-ache, nausea, abdominal cramps, tachycardia and rarely hypertension and hypotension. It should be avoided in infants and young children because of the risk of hyponatremia and in elderly with coronary artery disease(13). Antifibrinolytics The antifibrinolytic agents such as epsilon aminocaproic acid (Amicar, Hemostat) and tranexamic acid (Cyclokapron) have a special application in hemophilia. They inhibit plasminogen and thus prevent the lysis of the clot. They are useful to control bleeding during dental and oral surgery, thus reducing the need for replacement therapy. The dose of amino-caproic acid is 100 mg/kg (upto 6 g) every 6 hours intravenously and tranexamic acid (25 mg/kg orally or 10 mg/kg intravenously) every 6 to 8 hours. These agents should not be used to treat urinary tract hemorrhage as it stabilizes the clots which may result in clot colic. Minor infrequent side effects include nausea, vomiting, nasal congestion, watering of eye and skin rashes(14). Fibrin Sealant Fibrin sealant (fibrin glue, fibrin adhesives) have added a new dimension in the management of hemophilia. It imitates the final phase of hemostasis. It is available commercially as Beriplast-P and the product consists of fibrinogen, F XIII, aprotinin and thrombin. Fibrinogen is converted into fibrin on a tissue surface by the action of thrombin; the fibrin thus formed is then crosslinked by factor XIIIa to form a stable fibrin. Aprotinin is added to prevent excessively rapid fibrinolysis. These agents are useful for management of lacerated wounds, accessible bleeding sites and in dental extraction. Fibrin sealant has significantly reduced the need for factor replacement(15). References 1. ICMR Task Force. Collaborative Study on Hemophilia., New Delhi, Indian Council of Medical Research, 1990. 2. Choudhry VP, Kashyap R, Saxena R. Management of hemophilia in developing countries: An Indian experience. Hemophilia 1996; 2: 141-144. 3. Chandy M. Management of hemophilia in developing countries with available resources. Hemophilia 1995: 1 (Suppl 1): 44-48. 4. Hoyer LW. Hemophilia A. N Engl J Med 1994; 330: 38-47. 5. Buttler DB, Levine PH. Clinical manifestation and therapy of inherited coagulation factor deficiencies, Sazman EW. In: Hemostasis and Thrombosis: Basic Principles and Practice, 3rd edn. Eds. Colman RW, Hirsh J, Marder JW, Philadelphia, J.B. Lippincott, 1994; pp 169- 183. 6. Prevention and Control of Hemophilia. Memorandum from a Joint WHO/WFH (World Federation of Hemophilia) Meeting. Bull WHO 1991; 69: 17-26. 7. Gilchrist GS, Piepgras DG, Roskos RR. Neurological complications in Hemophilia. In: Hemophilia in the Child and Adult, 3rd edn. Eds. Hilgartner MV, Pochedly New York, Raven Press, 1989; pp 45-68. 8. Yue CP, Mann KS. The surgical management of intracranial hematomas in hemophiliac children. Child Nerv Syst 1986; 2: 5-9. 9. Webster WP, Rober HR, Penick GD. Dental care of patients with hereditary disorder blood coagulation. Med Treatment 1968; 5: 93-110. 10. Berntorp E. Methods of hemophilia care delivery. Regular prophylaxis versus episodic treatment. Hemophilia 1995; 1 (Suppl 1): 5-7. 11. Mannuci PM. Modern treatment of hemophilia from the shadows towards the light. Thromb Hemostat 1993; 70: 17-23. 12. Lusher JM. Considerations for the current and future management of hemophilia and its complications. Hemophilia 1995: 1: 2-10. 13. Aledort LM. Some aspect on the management of hemophilia. Thromb Hemostat 1995; 74: 440-443. 14. Sindet-Pedorsen S, Stenbjerg S. Efforts of local antifibrinolytic treatment with tranexaminc and in hemophiliacs undergoing oral surgery. J Maxillo Fac Surg 1986; 44: 703-707. 15. Gibble JW, Ness PM. Fibrin glue: The perfect operative sealant. Transfusion 1990; 30: 741-747. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||