|
Case Reports |
Indian Pediatrics 2000;37: 88-93 |
Dorfman-Chanarin Syndrome: A Rare Neutral Lipid Storage Disease |
| Milind S. Tullu,
Mamta N. Muranjan, Sushma U. Save, Chandrahas T. Deshmukh, Shaila R. Khubchandani* and
Burjor A. Bharucha+ From the Genetics
Division, Department of Pediatrics, Seth G.S. Medical College and K.E.M. Hospital, Parel,
Mumbai 400 012, India and *Department of Histopathology and Electron Microscopy, Jaslok
Hospital and Research Centre, 15, Dr. G. Deshmukh Marg, Mumbai 400 026, India. In 1953, Jordans described two brothers with progressive muscular dystrophy whose leukocytes contained fat vacoules(1). Similar abnormalities of the leukocytes (Jordan's anomaly) was reported by Rozenszajn in two sisters who also suffered from ichthyosis(1). Recently, several patients have been reported with non-membrane enclosed lipid droplets within many cell types, representing a unique inborn error of lipid metabolism which is clinically characterized by congenital ichthyosis, leukocyte vacoules and variable systemic involvement(2-11). To date, only 27 cases of this syndrome_the `Dorfman-Chanarin Syn-drome' (DCS) have been described world-wide(2). We report two more cases of this rare syndrome. Case Reports Case 1: A 6�-month-old male child born of non-consanguineous marriage with a normal perinatal period, presented with failure to thrive, delayed milestones, abdominal lump and pre-sence of rough skin noted since birth. There was history of an older female sibling being similarly affected who expired at the age of 6 months with jaundice and convulsions. This sibling had delayed milestones, ichthyosis and hepato-megaly (noticed at 1 month of age). On examination, the child had failure to thrive (weight: 4.5 kg_below 5th percentile), generalized ichthyosis, firm hepatomegaly of 8 cm and bilateral cataracts. Motor and social developmental quotient was three months. Other systemic examination was normal. A diagnosis of the `Dorfman-Chanarin Syndrome' (DCS) was considered based on the characteristic clinical features. Routine hematological work up including coagulation profile, renal chemistry, serum transaminases, GGT, alkaline phosphatase, serum protein profile and a-fetoprotein levels were normal. Fasting serum lipoprotein electrophoretic pattern revealed hyperlipidemia with elevated total serum cholesterol (153 mg/dl; normal: 30-110 mg/dl), Low Density Lipoprotein (LDL) cholesterol (111.8 mg/dl; normal: 10-50 mg/dl), Very Low Density Lipoprotein (VLDL) cholesterol (39.2 mg/dl; normal: 8-17 mg/dl) and serum triglycerides (196 mg/dl; normal: 30-86 mg/dl). High Density Lipoprotein (HDL) cholesterol (2 mg/dl); a-lipoprotein (17%), pre-b-lipoprotein (16%) chylomicrons (absent), b-lipoproteins (67%) and CPK-MM (23.2 units/L) were normal. Wright stained peripheral smear showed multiple refractory lipid vacoules in neutrophils and monocytes. His USG abdomen showed mild to moderate hepatomegaly with increased echo-genecity. The skeletal survey was normal. Opthalmological evaluation revealed bilateral cataracts with decreased lacrimation on Schirmer's test. Brainstem Evoked Response Audiometry (BERA) revealed mild hearing loss in the left ear and EMG-NC and EEG were normal. Light microscopic examination of the liver revealed altered architecture due to replacement by varisized nodules surrounded by a rim of fibrous tissue extending from the portal areas. The latter were infiltrated by aggregates of neutrophils and few lymphocytes. The hepatocytes showed diffuse fatty change. Above histology was consistent with neutral lipid storage disease and cirrhosis. Electron Microscopy (EM) of the liver tissue, skin and buffy coat smear confirmed the diagnosis of `Dorfman-Chanarin Syndrome'. Ultrathin sections of the liver and skin biopsy revealed that the cytoplasm and nuclei of the hepatocytes were distended with a large number of lipid droplets and plenty of lipid droplets were seen in the cytoplasm of keratinocytes of the prickle cell layer. Ultrathin sections of the buffy coat smears showed lipid droplets in cytoplasm of the neutrophils. The father's buffy coat smear (EM) also showed identical droplets in the neutrophils. However, they were not detected in the sample taken from the mother. The child was advised a moderate carbohydrate, low fat diet with medium chain triglycerides as the major source of fats. On follow up after 2 months, the liver size had regressed to 5 cm, ichthyosis had improved and there was a weight gain of 1 kg. Bilateral cata-ract removal with intraocular lens implantation had been carried out. There was no appreciable improvement in the milestones. Case 2: A 1�-year-old female child born of non-consanguineous marriage and an unevent-ful perinatal period, presented with dry skin lesions since birth and delayed milestones. Her 2 older male siblings were normal. On examination, there was failure to thrive (weight: 7.5 kg_below 5th percentile), general-ized ichthyosis (Fig. 1), firm hepatomegaly (3.5 cm with a span of 8 cm). Other than hypotonia, systemic examination was normal. Her motor and social developmental quotient was 6 months.
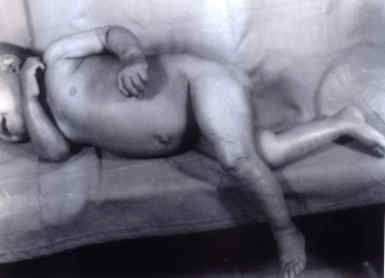
Fig1. Clinical photograph of case 2 demonstrating the non-bullouys ichthyosiform erythroderm, well seen on the lower limbs. Except for hypochromic, microcytic anemia, all hematological parameters and coagulation profile were normal. Light microscopic examination of her peripheral smear revealed vacoulated neutrophils, typical of DCS. Hepatomegaly with increased echogenecity of the liver was noticed on abdominal ultrasono-graphy. Her skeletal survey, fundoscopy, BERA, EMG-NC, EEG and MRI (Brain) were normal. Light microscopic examination of the liver revealed prominent fatty change of the hepatocytes and portal fibrosis. The hepatocyte cytoplasm stained positively with fat stains. Light microscopic examination of the skin biopsy was inconclusive. EM examination of the buffy coat revealed characteristic multiple fairly large oval lipid vacoules containing osmiophilic bar like structures, diagnostic of neutral lipid storage disease (Fig. 2). The child was advised a diet similar to that in Case 1.
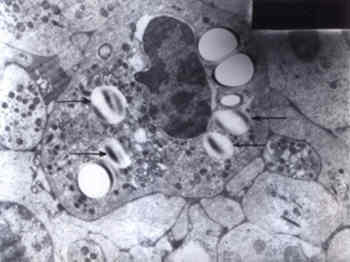
Fig. 2. Electron micrograph (� 6000)_The buffy coat reveals neutrophils showing large cytoplasmic vacoules containing osmiophilic material (arrows). Discussion Both the cases described above satisfy the major diagnostic criteria for the `Dorfman_ Chanarin syndrome', namely congenital ich-thyosis and systemic neutral lipid storage. The pathogenesis of the syndrome is poorly under-stood but appears to be related to perturbed intracellular triglyceride catabolism(2). Ultra-structural studies have shown mitochondrial involvement, suggesting a defect in mito-chondrial fatty acid oxidation(3,4). The syn-drome is inherited as an autosomal recessive trait(2-4). Presence of lipid droplets in basal and granulocytic layers of skin is a prominent histological finding(4) which was seen in the EM of skin biopsy in Case 1. Examination of the peripheral smear/buffy coat smear for the presence of characteristic pathognomonic lipid inclusions in the leukocytes is crucial for establishing the diagnosis of DCS(4). The non-membrane bound cytoplasmic inclusions primarily consist of neutral triglycerides(3,5). It has been emphasized that present day methods of automated peripheral blood counts do not detect the cytoplasmic vacuoles in the leuko-cytes and thus, microscopic examination of the peripheral blood smear is required for the diagnosis of DCS(4,6). Variable involvement of other organ systems in DCS has been described(2-4,6-9). Myopathy (with elevated serum muscle enzy-mes and abnormal EMG), mild neurologic impairment, ataxia, nystagmus, facial weakness, neurosensory deafness, developmental delay, cataracts, retinal dysfunction, mild ectropion (consequent to facial involvement by ichthyosis) and fatty liver (with/without mild elevation of hepatic enzymes) have been described(4). Growth retardation, gluten-sensitive entero-pathy, hepatosplenomegaly and diabetes melli-tus have been described in isolated reports(4,6). Apart from the skin and leukocytes, lipid inclusions can be demonstrated in the liver, gastro-intestinal tract (rectal, gastric and small bowel mucosa) and muscle fibres(3-6). Serum lipid profile has been inconsistent in various reports(3-7). Serum triglycerides, total choles-terol, LDL cholesterol and VLDL cholesterol were elevated in Case 1. Both the cases reported here had failure to thrive, developmental delay and hepatomegaly. In addition, Case 1 had bilateral cataracts and deafness. Deafness in DCS can develop at any age and can be progressive(4). Detection of heterozygotes by the presence of lipid droplets within the eosinophils of clinically unaffected parents has been empha-sized by previous workers(4,6). The father of Case 1 had lipid droplets in the neutrophils on EM examination of the buffy coat smear but the eosinophils were not prominently seen in the smear examined. The buffy coat examination of parents of Case 2 (light microscopy) was normal. The other disorder which shares some features with the DCS, namely, ichthyosis and organomegaly is the multiple sulfatase defi-ciency, but is distinguished from DCS by presence of coarse facial features, dysostosis multiplex and reduced levels of arylsulfatase A, B and C. Wolman's disease, a lysosomal storage disorder due to deficiency of acid lipase presents in the first year of life with massive hepato-megaly, failure to thrive and developmental regression and is differentiated from DCS by absence of ichthyosis, presence of adrenal calcification and a fatal outcome. The other disorders characterized by lipid storage in organs such as liver and muscle and myopathy are the disorders of carnitine metabolism, but again ichthyosis is not a feature of these diseases(4). Though rare, DCS should be considered in the differential diagnosis of congenital non-bullous ichthyosiform erythroderma. Treatment of DCS is essentially symptomatic(2,9). There are no guidelines regarding dietary management but a dietary modification with moderate carbohydrate and low fat diet with medium chain triglycerides may be useful. The life span is usually normal and the IQ may be normal or impaired(9). The visual and hearing deficits and the myopathy can be disabling(9). Parents should be counselled regarding a high risk of recurrence, which would be 25% since the disorder is inherited as an autosomal recessive trait. Prenatal diagnosis by fetal skin biopsy at 20 weeks has been performed(8). Also, prenatal detection may be possible by identification of characteristic lipid vacoules in amniocytes or leukocytes (obtained by fetal blood sampl-ing)(9); however this has not been studied as yet for its practical application. Acknowledgement We thank our Dean, Dr. (Mrs.) P.M. Pai for giving us permission to publish this case report. References 1. Rozenszajn L, Klajman A, Yaffe D, Efrati P. Jordan's anomaly in white blood cells: Report of case. Blood 1966; 28: 258-265. 2. Kaassis C, Ginies JL, Berthelot J, Verret JL. Dorfman-Chanarin Syndrome. Ann Dermatol Venereol 1998; 125: 317-319. 3. Angelini C, Philippart M, Borrone C, Bresolin N, Cantini M, Lucke S. Multisystem triglyceride storage disorder with impaired long chain fatty acid oxidation. Ann Neurol 1980; 7: 5-10. 4. Williams ML, Koch TK, O'Donnell JJ, Frost PH, Epstein LB, Grizzard WS, et al. Ichthyosis and neutral lipid storage disease. Am J Med Genet 1985; 20: 711-726. 5. Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral_lipid storage disease: A new disorder of lipid metabolism. Br Med J 1975; 1: 553-555. 6. Musumeci S D' Agata A, Romano C, Patane R, Cutrona D. Ichthyosis and neutral lipid storage disease. Am J Med Genet 1988; 29: 377-382. 7. Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 1974; 110: 261-266. 8. Judge MR, Atherton DJ, Salvayre R, Hilaire N, Levade T, Johnston DI, et al. Neutral lipid storage disease. Case report and lipid studies. Br J Dermatol 1994; 130: 507-510. 9. Williams ML. Storage Disease, Neutral lipid type. In: Birth Defects Encyclopedia. Eds. Buyse ML. USA, Blackwell Scientific Publications, Inc., 1990; pp 1607-1608. 10. Srebrnik A, Tur E, Perluk C, Elman M, Messer G, Ilie B, et al. Dorfman-Chanarin Syndrome. A case report and a review. J Am Acad Dermatol 1987; 17: 801-808. 11. Srebrnik A, Brenner S, Ilie B, Messer G. Dorfman_Chanarin Syndrome: Morphologic studies and presentation of new cases. Am J Dermatopathol 1998; 20: 79-85.
|