|
Brief Reports |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Indian Pediatrics 2000;37: 63-69 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
An Approach to Neurometabolic Disorders by a Simple Metabolic Screen |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sheffali Gulati, Meera Vaswani*,
Veena Kalra, Madhulika Kabra and Manjeet Kaur From
the Departments of Pediatrics and the *Division of Biochemistry, Department of Psychiatry,
All India Institute of Medical Sciences, New Delhi 110 029, India. Global surveys conservatively estimate the occurrence of inherited metabolic disorders in the range of three to four per thousand live born infants(1). In the multicentric Indian Council of Medical Research (ICMR) study, 4.9% of the genetic causes of mental retardation were due to metabolic disorders(2). The World Health Organization (WHO) estimated aminoacid disorders to account for approximately 10% of profoundly retarded children. Screening of mentally retarded children in India revealed that 0.5% to 2.4% of children had aminoacid disorders(3,4). Early diagnosis of metabolic disorders is desirable to reduce morbidity in cases where specific therapy is available. The spectrum of genetic counseling and prenatal diagnosis is rapidly expanding. Gene therapy is also on the anvil(5,6). Developing countries still suffer from paucity of facilities to investigate such patients. Detection of neurometabolic disorders is hampered by delay in early recognition of common phenotypes. The clinical features resemble more common early childhood illnesses like sepsis, failure to thrive, intrauterine growth retardation, recurrent vomiting and so forth(7). Lack of specificity of presenting clinical symptoms in these inherited metabolic diseases necessitates a large number of subjects to be probed by biochemical tests(7). The gamut of tests particularly useful in detecting and classifying neurometabolic disorders include blood gases, blood ammonia, urinary ketones and blood lactate(7,8). This communication presents our experience on screening for neurometabolic disorders by a simple metabolic screen. Subjects and Methods Children below two years of age attending the pediatric clinics or those admitted in the pediatric wards at the All India Institute of Medical Sciences between February 1994 to September 1995 were screened. Entry criteria included the presence of two or more of the following: seizures, encephalopathy, un-explained vomiting, developmental regression, mental retardation in the absence of major congenital anomalies, systemic signs of a neurometabolic disorder (ataxia, spasticity, cutaneous abnormalities, abnormal urinary or body odor, hair abnormalities, dysmorphic features or ocular abnormalities) and a positive family history(6). Thirty children entered the study. A predesigned clinical proforma was filled and features suggestive of neurometabolic disorders were carefully recorded. All children underwent a comprehensive metabolic screen which included blood and CSF [where possible] lactate, plasma aminoacidogram(9), urine aminoacidogram [organic aciduria where possible](9), urine for neurometabolic tests {ferric Chloride(10), dinitrophenylhydrazine [DNPH](11), reducing substances(12), nitro-prusside(13) and mucopolysaccharide [mps] spot test(12)}, urinary ketones, blood ammonia and arterial blood gases. Other tests included hemoglobin, peripheral blood counts, fasting blood sugar, serum electrolytes, calcium, hepatic and renal function tests. Neuroimaging, EEG (Electroencephalography) and muscle biopsy were done where indicated and possible. Estimation of blood and CSF lactate was standardized in our laboratory. Twenty five age matched normal children were taken as controls. Since CSF lactate estimation was not possible in healthy controls, literature norms were used(6,7). Arterial blood lactate was estimated in all the patients. CSF lactate was determined in ten and compared with blood lactate. Standard precautions like proper sampling and immediate processing were followed(5,14). Lactate estimation was done by the method of Marbach et al.(15). Patients were managed symptomatically and appropriate antiepileptic drugs instituted. Where investigations pointed towards specific disorders, treatment was instituted accordingly. All patients were regularly followed up. Results Patient characteristics included a male preponderance 23/30 (76.6%), positive family history in 12 (40%), with consanguinity in 3 (10%). Onset was in the first month of life in 13 (43.3%), within first year in 11 (36.6%) and second year in 6 (20%). Clinical presentation was acute in 15 (50%), chronic in 14 (46.6%) and intermittent in 1 (3.3%) which was precipitated by infection. Seizures were a dominant symptom being present in 27 (90%) followed by delayed development in 26 (86.6%) [out of which 18 (60%) had speech problems and 11 (36.6%) had regression of milestones], feeding problems in 23 (76.6%), failure to thrive in 18 (60%), encephalopathy in 13 (43.3%), overwhelming illness [encephalopathy, respiratory distress, seizures, septicemia, persistent vomitings] in 7 (23.3%), acidotic breathing in 5 (16.6%) and diarrhea in 5 (16.6%). The onset of seizures was within the first year of life in 21 of 27 (77.7%). Seizures were generalized in 15 (55.5%) [myoclonic in 2], partial in 8 (29.6%) [multifocal in 4] and mixed in 4 (14.8%). Most of the seizures were intractable to therapy and three-fourth of the patients required two or more anti-epileptic drugs despite which, 11 of 27 (40.7%) remained uncontrolled. Examination revealed microcephaly in 16 (53.3%), failure to thrive in 13 (43.3%), dysmorphic facies in 10 (33.3%), abnormal urine or body odor in 5 (16.6%), hair changes [alopecia, light colored hair] in 5 (16.6%), dermatologic changes [evanescent erythematous papular lesions, depigmented macules] in 2 (6.6%) and deafness in 2 (6.6 %) patients. Hepatomegaly �3 cm was present in 7 (23.3%) and splenomegaly in 4 (13.3%) patients. Central nervous system abnormalities included optic atrophy in 9 (30%), cherry red spots in 2 (6.6%), extrapyramidal symptoms in 7 (23.3%), long tract signs in 19 (63.3%) and hypotonia in 10 (33.3%) patients. The blood ammonia levels ranged from 7 to 980 mg/dl, levels >80 mg/dl were reconfirmed by repeat tests. Significant elevation (>200 mg/dl) was observed in four patients, while ten had borderline rise (80-200 mg/dl) which was explained by intercurrent antiepileptic drug therapy especially sodium valproate. Homocystinuria was diagnosed in one patient by increased homocystine on plasma aminoacidogram. DNPH test was positive in the patient with biotinidase deficiency. Nitro-prusside test was positive in the patient with homocystinuria and was falsely positive in another patient on nebulized N-acetyl cysteine. Electroencephalogram [EEG] were abnormal in 72.3% and were unrelated to the seizure type. Cranial computed tomography [CT] was abnormal in 16/26 (61.5%) with cerebral atrophy in 7 (26.9%), basal ganglia abnormalities in 3 (11.4%), dysmyelination in 2 (7.6%), hypodense areas in 2 (7.6%), cerebral edema in 1 (3.8%) and widespread ischemic damage in 1 (3.8%). Cranial magnetic reso-nance imaging (MRI) done in 6 of 30 (20%) patients revealed additional findings in three patients suggestive of mitochondrial disorder (n=1), migration defect (n=1) and cerebral atrophy (n=1). It confirmed CT findings in two patients having leukodystrophy (n=1) and Leigh's disease (n=1). Based on four screening metabolic tests instituted(7,8) a probable diagnosis could be arrived at in 12/30 (40%) patients (Table I). The commonest disorder was organic aciduria followed by cerebral lactic acidosis, urea cycle disorder, mitochondrial disorder and homo-cystinuria type II. Table I__Clinico-Biochemical Correlation in Patients with Neurometabolic Disorders
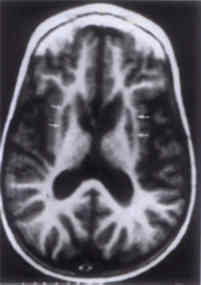
BM_Bone marrow; DD_Developmental delay; E_Encephalopathy; FBS_Fasting blood sugar; OA_Optic atrophy; S-Seizure Four patients with elevated blood lactate(16) could be categorized as follows. Patient 1 had encephalopathy, uncontrolled seizures, delayed development, alopecia, erythematous skin rash, hyperammonemia and ketoacidosis suggestive of organic aciduria. Urinary organic acid estimation revealed increased urinary excretion of 3-hydroxyisovaleric acid, 3-methylcrotonic acid and 3-methylcrotonylglycine which was consistent with biotinidase deficiency. Optic atrophy, significant hyperammonemia, hypo-glycemia, acidosis and absence of ketonuria with progressive deterioration indicated a fatty acid oxidation defect or organic aciduria (HMG-CoA lyase deficiency) in Patient 2. Patient 3 had progressive neurologic regression and acidosis. CT showed bilaterally symmetrical basal ganglia hypodensities and MRI was suggestive of Leigh's disease [areas of signal hypointensity in basal ganglia on T1-weighted images (Fig. 1)]. Patient 4 had lactic acidosis and seizures. This patient could not be categorized further.
Fig. 1. Areas of signal hypointensity in putamen bilaterally as well as in the head of the caudate nucleus on T1-weighted images (Leigh's disease). Three patients [cases 5, 6 and 7] with elevated CSF lactate, normal or mildly elevated blood lactate and minimal or no systemic acidosis were diagnosed as cerebral lactic acidosis(17). All had seizures, optic atrophy [Patient 5] and abnormal body and urine odor [Patient 7]. Isolated significant hyperammonemia suggested a urea cycle disorder in two [Patients 8 and 9]. Patient 10 had encephalopathy, ketoacidosis with hypoglycemia suggesting an organic aciduria. Another patient with encephalopathy [Patient 11] had ketoacidosis and basal ganglia infarcts. Possibilities of organic aciduria and mitochondrial disorder were entertained. Patient 12 was diagnosed as homocystinuria type II based on positive urine nitroprusside test, raised plasma homocystine and megaloblastic changes in the bone marrow. All patients were managed symptomatically. The probable metabolic defect identified helped in instituting specific therapeutic interventions in five patients in the form of biotin [biotinidase deficiency (Patient 1)](18), sodium benzoate and dietary protein restriction [fatty acid oxidation defect/organic aciduria (Patient 2), urea cycle disorder (Patient 8)] (5,18), thiamine [Leigh's disease (Patient 3)](5,19) and Vitamin B12 and folate [homocystinuria type II (Patient 12)](19) with favorable results. Four (13.3%) patients died during follow up. Discussion The initial step in the evaluation of any sick neonate, and clearly, the most important one, is a thorough clinical assessment including family history(20). Neurometabolic disorders are commonly transmitted as autosomal recessive traits(1,5,19), the male predominance is un-explained except for a possible gender bias in hospitalization of sick children. A positive family history and consanguinity were the most important pointers. This is in agreement with literature(20). An early onset in the majority of patients is corroborated by earlier reports. Burton et al. reported that manifestation of inherited metabolic disorders whether overt, life threatening or subtle was generally present in the neonatal period(20). The need for screening sick neonates is stressed. The profile of clinical features observed was similar to that reported in literature(7,20). The most prevalent features reported include mental retardation followed by seizures and ataxia(21). In the current study, seizures, developmental retardation, feeding problems and failure to thrive were predominant symptoms. Seizures are more likely to be uncontrolled in metabolic disorders. None of the clinical features alone or in combination with uncontrolled seizures pointed towards a metabolic disorder. This is in total agreement with literature, which states that clinical features as nonspecific(6), though the small sample size could be partly contributory. Hence a metabolic screening is essential in sick neonates, especially with an unidentified cause or in absence of fever. A high index of suspicion is essential to identify neurometabolic disorders at an appropriate age when intervention is likely to be useful. There is paucity of metabolic screening facilities in developing countries. A battery of four simple tests namely arterial blood gases, blood ammonia, urinary ketones and blood lactate(7,8) in tertiary health care centers is advocated. These tests have considerable degree of overlap, for example, lactate levels may be elevated or normal in organic acidurias. Hyper-ammonemia may be present in some organic acidurias. Some conditions with lactic acidosis may have hyperammonemia. Hence, if there is only ketosis then maple syrup urine disease is the likely diagnosis. Ketoacidosis with or without elevated lactate and hyperammonemia point towards organic aciduria. Elevated lactate along with ketoacidosis with or without hyper-ammonemia is seen in lactic acidosis. If hyper-ammonemia is an isolated finding it points towards a urea cycle disorder. In case all of the four tests are negative, non-ketotic hypergly-cemia or peroxisomal disorders are likely(7). Patients with "cerebral lactic acidosis" show neurological symptoms, elevated levels of lactate in CSF, little or no systemic acidosis and levels of lactate in blood so slightly elevated that they would be overlooked. Lactate elevation confined to CSF and brain has been described in biotinidase deficiency and in some mito-chondriopathies(17,22,23). Forty per cent of patients could be broadly classified. A precise diagnosis requires specific tests(7), which were not available and only one patient could be diagnosed. Organic acidurias were found to be most common followed by cerebral lactic acidosis, urea cycle disorders, mitochondrial disorder and homocystinuria type II which is in consonance with that reported in literature(7,8). Screening studies are recom-mended on a larger scale. Based on this pilot study, it is recommended that in developing countries where economic constraints limit these facilities, there is a need to provide simple tests to broadly classify and identify neurometabolic disorders. They are not uncommon in infants with acute illness with nonspecific symptoms. References 1. Chalmers RA, Watts RWE, Lawson AM. A comprehensive screening method for detecting organic acidurias and other metabolic diseases in acutely sick infants and children. Ann Clin Biochem 1977; 14: 149-156. 2. ICMR Collaborating Centres and Central Co-ordinating Unit. Multicentric study on genetic causes of mental retardation in India. Indian J Med Res 1991; 94: 161-169. 3. Ambani LM, Patel ZM, Dhareshwar SS, Krishnamurthy DS, Moghe MS, Mulye VR, et al. Clinical, biochemical and cytogenetic studies in mental retardation. Indian J Med Res 1984; 79: 384-387. 4. Kaur M, Das GP, Verma IC. Inborn errors of aminoacid metabolism in North India. J Inherit Metab Dis 1994; 17: 1-14. 5. Aicardi J, Ogier H. Metabolic diseases. In: Diseases of the Nervous System in Childhood, 1st edn. Ed. Aicardi J. New York, Cambridge University Press, 1992; pp 379-517. 6. Menkes JH. Metabolic diseases of the nervous system. In: Textbook of Child Neurology, 5th edn. Ed. Menkes JH. Maryland, Williams and Wilkins, 1995; pp 29-151. 7. Carballo EC. Detection of inherited neuro-metabolic disorders_A practical clinical approach. Pediatr Clin North Am 1992; 39: 801-820. 8. Saudubray JM, Ogier H, Bonnefont JP. Clinical approach to inherited metabolic disease in the neonatal period: A 20-year survey. J Inherit Metab Dis 1989; 12 (Suppl 1): 25-41. 9. Ireland JT, Read RA. A thin layer chromato-graphic method for use in neonatal screening to detect excess amino acidaemia. Ann Clin Bio-chem 1972; 9: 129-132. 10. Carpenter GG, Auerbach VH, Digeorge AM. Ferric chloride test. Pediatr Clin North Am 1968; 15: 554-555. 11. Centerwall W, Chinnock R, Pusavat A. Phenyl-ketonuria: Screening programme and testing methods. Am J Pub Hlth 1960; 50: 1667-1677. 12. Buist NRM. Set of simple sideroom urine tests for detection of inborn errors of metabolism. Brit Med J 1968; 2: 745-749. 13. Spaeth GL, Barber GW. Prevalence of homo-cystinuria among the mentally retarded: Evaluation of a specific screening test. Pediatrics 1967; 40: 586-589. 14. Robinson BH. Lactic acidemia. In: The Meta-bolic Basis of Inherited Disease, 6th edn. Eds. Scriver CR, Beaudet AL, Sly WS, Valle D. New York, McGraw-Hill, 1989; pp 869-888. 15. Marbach EP, Weil MH. Rapid enzymatic measurement of blood lactate and pyruvate. Clin Chem 1967; 13: 314-325. 16. Panteghini M, Pagani F. Biologial variation of lactate and pyruvate in blood. Clin Chem 1993; 39: 908. 17. Hoffman GF, Augenstein WM, Stockler S, Surtees R, Rating D, Nyhan WL. Physiology and pathophysiology of organic acids in cerebro-spinal fluid. J Inherit Metab Dis 1993; 16: 648-669. 18. Levy HL. Nutritional therapy for selected inborn errors of metabolism. J Am Coll Nutr 1989; 8 (Suppl): S54-S60. 19. Swaiman KF. Aminoacidopathies and organic acidemias resulting from deficiency of enzyme activity. In: Pediatric Neurology - Principles and Practice, 2nd end. Ed. Swaiman KF. Missouri, Mosby Publications, 1994; pp 1195-1232. 20. Burton BK. Inborn errors of metabolism. The clinical diagnosis in early infancy. Pediatrics 1987; 79: 359-369. 21. Hagberg B. Neurometabolic disorders in infancy, childhood and adolescence. Acta Neurol Scand 1967; 43 (Suppl 31): 13-19. 22. Fois A, Cioni A, Balestri P, Bartilini G, Baumgartner R, Bachmann C. Biotinidase deficiency. Metabolites in CSF. J Inher Metab Dis 1986; 9: 284-285. 23. Brown GK, Haan EH, Kirby DM, Scholem RD, Wraith JE, Rodgers JG, et al. "Cerebral" lactic acidosis: defects in pyruvate metabolism with profound brain damage and minimal systemic acidosis. Eur J Pediatr 1988; 147: 10-14. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||