Shubha R. Phadke
Ashutosh Gupta
Jagannath Pahi
Amita Pandey
Prachi Gautam
S.S. Agarwal
From the Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226 014.
Reprint requests: Dr. Shubha R. Phadke, Assistant Professor, Department of Medical Genetics, San jay Gandhi Postgraduate. Institute of Medical Sciences, Raebareli Road, Lucknow 226 014, India.
Manuscript received: May 26,
1998;
Initial review completed: June 19, 1998;
Revision accepted: July 27, 1998.
Osteopetrosis encompasses a heterogeneous group of inherited disorders of the
skeleton caused by defect in
bone
resorption
by the osteoclasts. It is characterized by in- creased skeletal mass and bone
density as detected radiologically. The autosomal dominant (AD) form of osteopetrosis is usually asymptomatic. It is diagnosed incidentally or may exhibit mild symptoms in late childhood or adult life, but is compatible with long term survival. In contrast, the autosomal recessive (AR) form manifests with severe symptoms of abnormal bone remodelling,
deficient hematopoiesis and neurological impairment in infancy, resulting in markedly reduced life span. It is also referred to as malignant recessive osteopetrosis. The existence of a clinically intermediate form has also been reported(1). AR osteopetrosis is a rare disorder, the incidence being 1 in 200,000(2). Here we present our experience of AR osteopetrosis. The currently available therapeutic options are also reviewed.
Patients
We have come across six patients of AR osteopetrosis in our OPD during a period of five years. The clinical features and laboratory
findings of these patients are summarized in Table I. The median age of presentation was 5.5 months (range
-
3 months to 1year). There were 5 males and 1 female. Four patients were born to
Muslim parents, with history of parental consanguinity (first
cousins) in two families. 111 one patient (Case 2) there was history of two similarly affected siblings. The medical records of the siblings, who died at the age of 4 months and 8 months, showed that they had anemia, hepatosplenomegaly
and roving eye movements. One was diagnosed to have congenital rubella infection and the other juvenile chronic myeloid leukemia (hemoglobin of 5.4 g/dl and total leukocyte count of 18,0001 mm3 with 20% blast cells) Incidentally chest X-rays of both were available and showed in- creased bone density of ribs and spine and bone in bone appearance in humeri
suggesting that probably they were also suffering from AR osteopetrosis. Two patients' were born to non-consanguineous Hindu couples.
TABLE I - Clinical Features and Laboratory Findings of
Patients with Osteopetrosis
|
Case |
1 |
2 |
3 |
4 |
5 |
6 |
| Age at
Presentations (mo) |
3 |
4 |
5 |
6 |
6 |
12 |
| Sex |
Male |
Male |
Male |
Female |
Male |
Male |
|
Religion |
Muslim |
Muslim |
Hindu |
Hindu |
Muslim |
Muslim |
|
Referral diagnosis |
Hemolytic
anemia |
Thalassemia
major |
Hepatospleno-megaly with anemia |
Subdural hematoma with anemia |
Hepatospl-enomegaly
|
Thalassemia
Major
|
|
Symptoms |
Pallor,
distension of abdomen |
Pallor,
distension of abdomen |
Fever,
pallor |
Increasing head size |
Bulging
head, nasal block |
Pallor,
increasing head size |
|
Consanguinity |
+ |
+ |
- |
- |
- |
- |
| Family
history |
- |
+ |
- |
- |
- |
- |
|
Prominent forehead |
- |
+ |
+ |
+ |
+ |
- |
|
Macrocephaly |
- |
- |
- |
+ |
+ |
- |
|
Depressed nasal bridge |
- |
+ |
+ |
+ |
+ |
- |
| Anemia |
++ |
+++ |
+++ |
+++ |
+++ |
+ |
| Nasal
blockade |
+ |
+ |
- |
? |
+
Epistaxis |
+ |
| Liver
(cm) |
5 |
6 |
4 |
5 |
6 |
2 |
| Spleen
(cm) |
2 |
10 |
4 |
8 |
6 |
10 |
| Optic
atrophy |
- |
- |
+ |
+ |
+ |
- |
|
Hemoglobin (g/dl) |
7.4 |
6.4 |
7.2 |
5.0 |
6.1 |
8.5 |
| TLC (x103/µL) |
9.8 |
58 |
15.2 |
12 |
45 |
44 |
|
Platelets (x106/µIL) |
0.61 |
0.64 |
0.91 |
0.68 |
0.80 |
1.26 |
|
Premature myeloid cells in peripheral smear (%) |
6 |
32 |
15 |
11 |
16 |
18 |
| Normoblasts (/100 WBC) |
2 |
35 |
19 |
25 |
45 |
7 |
| Serum
calcium (mg/dl) |
9.6 |
9.8 |
9.2 |
10.0 |
9.5 |
9.3 |
| Serum
inorganic phosphours (mg/dl) |
5.6 |
5.4 |
6.0 |
5.8 |
5.8 |
5.1 |
| Serum alkaline
phosphatase (U/L) |
264 |
1535 |
869 |
158 |
188 |
419 |
| Skeletal X-rays |
|
|
|
|
|
|
| a.
Increased bone density |
+ |
+ |
+ |
+ |
+ |
+ |
| b. Bone in bone
appearance |
+ |
+ |
+ |
+ |
+ |
+ |
| Cranial
CT scan |
Not
done |
Not done |
Normal |
Subdural hematoma |
Hydrocep-halus, aqu-eductal stenosis |
Not done |
| outcome |
Lost to
follow-up |
Died |
Lost to
follow-up |
Died |
Lost to follow-up |
Lost to follow-up |
Increasing pallor and listlessness was the
most common initial symptom (n=4), other
symptoms being distension of abdomen, fever, increasing head size and nasal block. Five patients had more than one symptom.
On examination, all patients had anemia and hepatosplenomegaly. Nasal obstruction was seen in four patients one of whom also had epistaxis. Ophthalmological examination revealed optic atrophy in three. Cranial CT scan done in these three patients revealed normal anatomy in one, hydrocephalus with aqueductal stenosis in one and subdural hematoma in one. The latter patient (Case 4) had presented to the Department of Neurosurgery at the age of six months with
complaints of increasing head size and delayed milestones. She was found to be pale, had hepatosplenomegaly, macrocephaly and optic atrophy. The subdural hematoma was evacuated surgically. Skeletal X-rays clinch- ed the diagnosis of osteopetrosis.
The hemoglobin values were low in all the patients (6.8±1.22 g/dl). Thrombocytopenia and leukocytosis were seen in five patients each. Peripheral smear showed the presence of immature leukocytes (myelocytes and metamyelocytes 6-32%) and normoblasts (2-45/100 WBC) in all the patients. Serum calcium and inorganic phosphorus were normal in all. Serum alkaline phospha1ase was elevated in two.
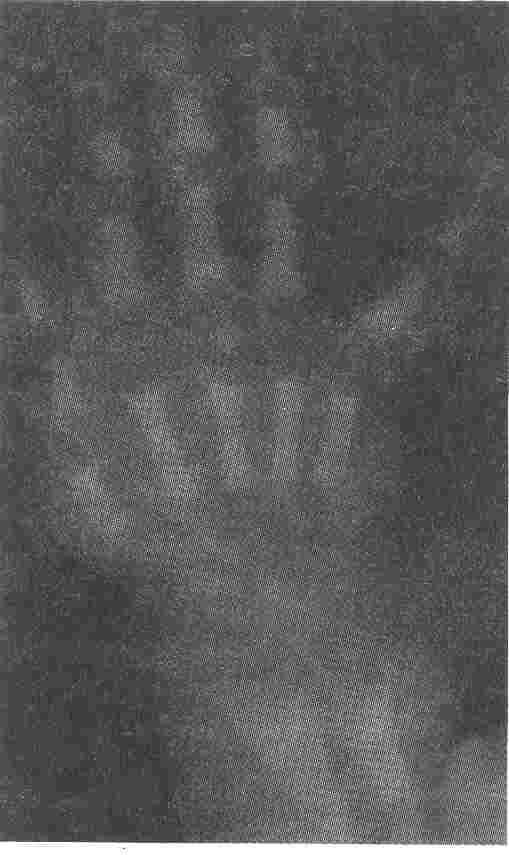
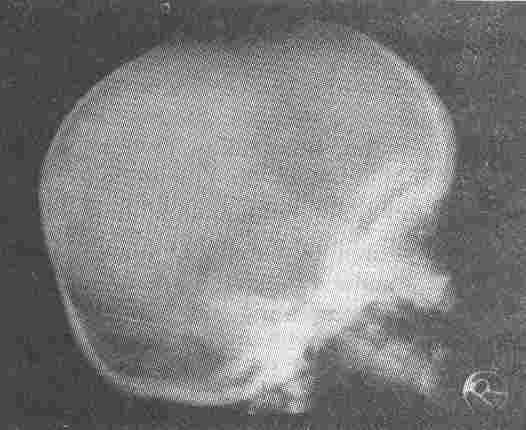
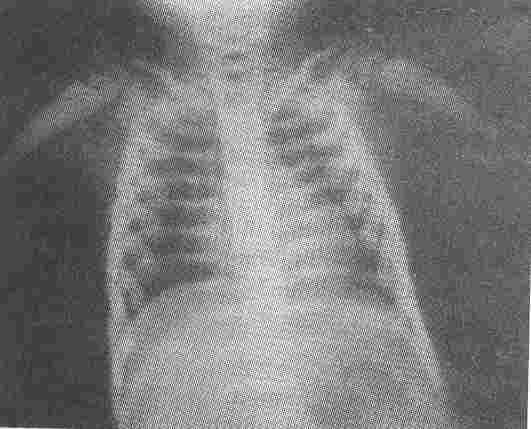
The skeletal X-rays (Figs. 1-3) showed generalized increase in bone
density, widening and modelling deformity of metaphyses of long bones and "bone in bone" appearance in the phalanges, long bones and pelvic bones of all the patients and confirmed the diagnosis of osteopetrosis.
Discussion
Autosomal recessive osteopetrosis is a rare disorder and reports of large groups of patients are scarce. Loria-Cortes' detailed a cr9ss-sectional study of 26 patients from Costa Rica(3) and Gerritsen's retrospective longitudinal study of 33 patients(4) have summarized the clinical picture and natural course of the disease.
 |
|
Fig. 1. Radiograph of hands showing increased. bone density and bone in bone appearance. |
The commonest presenting symptom in our patients was pallor and listlessness. Other authors have reported nasal obstruction(3) and visual impairment(4) as the initial symptoms. Nasal obstruction was present in four of our patients although it was the primary complaint in only one.
Our findings are in agreement with those -reported in the above studies: Most of the
children present before 6 months of age with anemia, hepatosplenomegaly and loss of vision either alone or in combination. The hematological. manifestations are due to obliteration of marrow cavity leading to myelophthisic anemia with reticulocytosis, normoblastosis, and leukocytosis of immature myeloid series. Hepatosplenomegaly develops because of extramedullary hematopoiesis. Ensuing hypersplenism leads to thrombocytopenia, leukopenia and hemolytic anemia(5). The risk of developing hematological impairment in the first year of life is about 75% and its onset within 3 months of life is indicative of a poor outcome(4).
 |
|
Fig. 2. Increased density of bones of the base of the skull. |
 |
|
Fig. 3. Dense bones and modelling deformity of metaphyses of humori. |
Visual impairment due to optic nerve encroachment is a common feature at presentation. Surgical decompression of optic nerve may restore sight in some cases{6,7). Cumulative risk of developing a visual defect in the first year of life is about 75%. Combined early visual and hematological impairment is very significantly associated with a poor outcome(4).
Other features like macrocephaly, nystagmus, exophthalmos, strabismus, petechiae, nasal block, psychomotor retardation, failure to thrive and convulsions may be present(3). One of our patients had hydrocephalus due to aqueductal stenosis.
The characteristic radiological feature of osteopetrosis is generalized sclerosis of bone. Other radiological changes observed are splaying of metaphyses and rib ends, vertical or transverse metaphyseal lines, bone in bone appearance, sandwich appearance of vertebral bodies (rugger jersey spine), non-visualization of marrow cavity, pathological fractures, curvature of bones, osteomyelitis and sun burst appearance of skull vault(8). AR osteopetrosis can manifest in utero with multiple fractures(9) and may cause non- immune hydrops fetalis(10).
The natural course of the disease results in survival of about 30% of patients at six years of age. Some may live till 2nd or 3rd
decade but the quality of life is mostly
poor(4).
Bone marrow transplantation (BMT) pro- vides the only curative treatment for AR
osteopetrosis. Recipients of HLA identical BMT have been reported to have 5 year survival of 79% while survival falls to 38% with one HLA mismatch and to 13% with a com- plete HLA mismatch(11).
Clinical outcome with paratharmone, calcitrol and prednisone therapy has been found to be inconsistent and variable(12-14). Recently, use of interferon gamma has been found to decrease the rate of infection and transfusion requirements after 24 months of therapy(15).
Though rare, AR osteopetrosis should be considered in the differential diagnosis of an infant or young child presenting with anemia
and hepatosplenomegaly otherwise he/she may be wrongly diagnosed as leukemia or other hemolytic anaemia. The diagnostic test, i.e., skeletal radiography is simple and readily available. An accurate diagnosis is essential in view of availability of curative treatment (BMT) and for genetic counselling as the risk of recurrence in siblings is 25%.
|
1.
Beighton P, Hemersma H, Cremin BJ. Osteopetrosis in South Africa:
The benign, lethal and intermediate forms. S Afr Med J .1979; 55: 659-665.
2.
Johnston CC, Lavy N, Lord T, Vellios F, Merritt AD, Deiss WP. Osteopetrosis: A clinical, genetic, metabolic, morphologic study of the dominantly inherited, benign form. Medicine 1968; 47: 149-167.
3.
Loria-Cortes R, Quesada-Calvo E, Cordero-Chaverri C, Osteopetrosis in children: A report of 26 cases. J Pediatr 1977;91: 43-37.
4.
Gerritsen EJA, Vossen JM, Van Loo IHG, Hermans J, Helfrich MH, Griscelli C, Fischer A. Autosomal recessive osteopetrosis: Variability of findings at diagnosis and during the natural course. Pediatrics 1994; 93: 247- 253.
5.
Alber BP, Young NS. Bone marrow failure syndromes. In: Hematology of Infancy and Childhood, 4thedn. Eds. Nathan DG, Oski FA. f'ennsylvania, W.B. Saunders Company, 1993; pp 216-316.
6.
AI-Mefty 0, Fox JL, AI Rodhan N, Dew JH. Optic nerve decompression in osteo-petrosis. J Neurosurg 1988; 68: 80-84.
7.
Haines SJ, Erickson DL, Wirtschafter JD. Optic nerve decompression for osteopetrosis in
early childhood. Neurosurgery 1988; 23: 470- 475.
8.
Lachman RS, Osteopetrosis. In: Radiology of
Syndromes, Metabolic Disorders and Skeletal Dysplasias, 4th edn. Eds. Taybi H, Lachman RS, Missouri, Mosby Year Book Inc., 1996; pp 886-891.
9.
EI Khazen N, Faverly D, Vamos E, Van Regemorter N, Flament DJ, Carton, et al. Lethal osteopetrosis with multiple fractures in utero. Am J Med Genet 1986; 23: 811- 819.
10.
Mathur BP, Karan S. Non immune hydrops fetalis due to osteopetrosis congenita, Indian Pediatr
1984; 21: 651-653.
11.
Gerritsen EJA, Vossen JM, Fasth A, Friedrich W, Morgan G, Padmos A, et al. Bone marrow transplantation for autosomal recessive osteopetrosis: A report from the working party on inborn errors of the European Bone Marrow Transplantation Group. J Pediatr
1994; 125: 896-901.
12. Van Lie Peters EM, Aronson DC, Dooren LJ.
Failure of calcitriol treatment in a patient with malignant ostebpetrosis. Eur J Pediatr 1993; 152: 818-821.
13.
Reeves Jp, Huffer WE, August CS, Hathway WE, Koerper M, Walters CE. The hematopoietic effects of prednisone therapy in four
infants with osteopetrosis. J Pediatr 1979; 94:
210-214.
14.
Glorieux FH, Petti for JM, Marie PJ, Delvin EE, Travers R, Shepard N. Induction 'of bone resorption by parathyroid hormone in congenital malignant osteopetrosis. Metab Bone Dis Relat Res 1981; 3: 143-150.
15.
Key LL Jr., Rodriguiz RM, Willi SM, Wright NM, Hatche HC,. Eyre DR, et al. Long term treatment of osteopetrosis with recombinant human interferon gamma. New Engl J Med 1995; 332:1594-1599.
|
