Inborn errors of immunity or primary immune
deficiency disorders (PIDDs) occur with a frequency of 1 in 5000 to 1 in
1000 [1], and are frequently misdiagnosed resulting in avoidable
morbidity and mortality [2]. Diagnostic tests and hematopoietic stem
cell transplant (HSCT) are not uniformly accessible [3].
Government Medical College, Kozhikode, a tertiary
care hospital in Kerala, and CSIR Institute of Genomics and Integrative
Biology, Delhi have been conducting a program on primary immune
deficiency disorders over the last five years. Although HSCT is often
the only curative option, we are dependent on centers outside the state.
The study was designed to document the clinical characteristics of
children who underwent HSCT for an inborn error of immunity.
Hospital records of children with PIDDs who attended
the immune deficiency clinic from June, 2015 to May, 2020 were obtained
and data of those who underwent HSCT were analyzed. Only children who
had completed at least 3 months post-HSCT were included. Variables
studied included age at onset diagnosis and at HSCT, gender,
relationship with stem cell donor, time since HSCT and diagnostic
genetic or phenotypic marker. Quantitative variables were entered on an
Excel data sheet and frequency and associations calculated using the
statistical package Epi Info (version 7.2.3.1).
HSCT was performed in 13/67 (19.4%, 11 boys). The
indications included Wiskott-Aldrich syndrome (4, 30.8%), and leukocyte
adhesion deficiency, severe combined immune deficiency, and X-linked
agammaglobulinemia in two each (15.4%) congenital neutropenia Fanconi
anemia, and hyper IgM syndrome were diagnosed in one child each. The
median (IQR) age at diagnosis of children who underwent HSCT was 14
months (first quartile, III quartile). The median (IQR) age at HSCT was
27.5 (first quartile, III quartile) months and the median (IQR) interval
between diagnosis and HSCT was 7 (first quartile, III quartile) months.
Recurrent pneumonia was the commonest presenting feature in 7 (54%)
children, followed by frequent skin and soft tissue infections in 6
(46%) and recurrent otitis media in 4 (30.8%). Frequent abscesses,
recurrent diarrhea and bleeding were presenting features in 2 (15%)
children each. HSCT was done in an asymptomatic child with Fanconi
anemia after his elder sister succumbed to the same disease.
Of the 13 children who underwent HSCT, 9 (69%)
children had a matched sibling donor and 2 children each (15%) had
matched unrelated donor transplants (MUDs) [4] and haploidentical stem
cell transplants. Reduced intensity conditioning (RIC) [5] with
treosulfan and fludarabine was used and 12 children had sustained
engraftment. There was one graft rejection with autologous
reconstitution, and a second HSCT resulted in sustained engraftment.
Post-HSCT complications included bacterial sepsis, cytomegaloviral
reactivation, steroid-induced hypertension and graft versus host
disease. There was no mortality and the mean duration of post-transplant
event-free survival was 25.1 months.
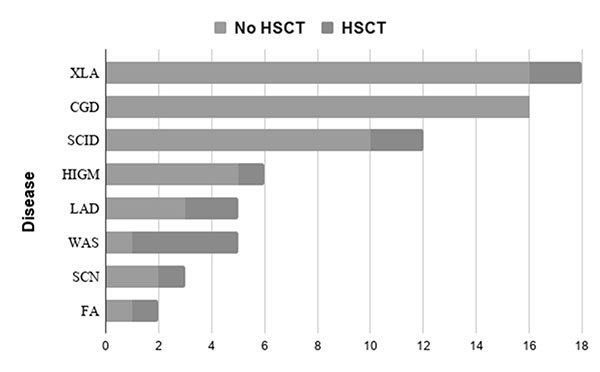
SCN: Severe congenital neutropenia; FA:
Fanconi anemia; HIGM: Hyper-IgM syndrome; LAD1: Leukocyte
adhesion deficiency; SCID: Severe combined immune deficiency;
WAS: Wiskott–Aldrich syndrome; XLA: X-Linked agammaglobulinemia.
|
|
Fig. 1 Number of patients with primary immune deficiency
disorders who underwent hematopoietic stem cell transplants
(HSCT).
|
HSCT was performed for 2 (15%) children with XLA.
Although this is not the standard treatment, but it has been found to be
a feasible option where availability and cost of immunoglobulin
replacement therapy are limiting factors and parents are not keen on
lifelong replacement [6].
The median interval between onset of symptoms to
diagnosis was 9 months. This emphasizes the need to improve awareness
among pediatricians [2]. The mean interval between diagnosis and
HSCT was 40.9 months, accounting for the high mortality. Improved
outcomes are described with HSCT before 3.5 months of age before onset
of infectious complications [7,8]. The youngest child who underwent HSCT
in this series was 5 months.
The outcome of HSCT for children with matched
unrelated donors (MUDs) and haploidentical donors has improved globally
[4,9] both children in this series had good outcomes. Limitations of the
study include the small sample size and the variable time since HSCT
with possible recall bias.
The main stumbling blocks to wider use of HSCT remain
the cost and non-availability of suitable donors. National rare disease
policy addressing the major concerns of affected families would be the
way forward. Awareness regarding PIDDs should be rapidly scaled up,
donor registries expanded and government funding streamlined. A newborn
screening program would help to reduce mortality.
Acknowledgements: Dr. Dhanasooraj, Scientist,
MRU, Govt. Medical College, Kozhikode; Dr. Ajith Kumar VT and Dr. MP
Jayakrishnan, Department of Pediatrics, Government Medical College,
Kozhikode; Athulya EP, Junior Research Fellow; and Abhinav Jain and Dr.
Sridhar Sivasubbu at the CSIR - Institute of Genomics and Integrative
Biology, Delhi.
Ethics clearance: GMCKKD/RP2020/IEC/428; dated
May 29, 2017.
Contributors: GMG: conceptualization of the
study, data analysis and writing the paper. RR and RU oversaw the
work-up and procedure for HSCT; VS: did the genetic work up for the
patient. All authors approved the final draft of the paper.
Funding: Science and Engineering Research Board,
Delhi, and Foundation for Primary Immune Deficiency Diseases (FPID);
Competing interesst: None stated.
REFERENCES
1. Tangye SG, Al-Herz W, Bousfiha A, et al. Human
Inborn Errors of Immunity: 2019 Update on the Classification from the
International Union of Immunological Societies Expert Committee. J Clin
Immunol. 2020;40:66-81.
2. Kapoor N, Raj R. Hematopoietic stem cell
transplantation for primary immune deficiency disorders. Indian J
Pediatr. 2016;83:450-54.
3. Jindal AK, Pilania RK, Rawat A, Singh S. Primary
immunodeficiency disorders in India – A situational review. Front
Immunol. 2017; 8:714.
4. Dalal I, Reid B, Doyle J, et al. Matched unrelated
bone marrow transplantation for combined immunodeficiency. Bone Marrow
Transplant 2000;25:613-21.
5. Rao K1, Amrolia PJ, Jones A, et al. Improved
survival after unrelated donor bone marrow transplantation in children
with primary immunodeficiency using a reduced-intensity conditioning
regimen. Blood. 2005;105:879-85.
6. Ikegame K,Imai K, Yamashita M, H, et al.
Allogeneic stem cell transplantation for X-linked agamma-globulinemia
using reduced intensity conditioning as a model of the reconstitution of
humoral immunity. J Hematol Oncol. 2016; 9:9.
7. Szabolcs, M. Cavazzana-Calvo, A. Fischer, Veys P.
Bone marrow transplantation for primary immunodeficiency diseases.
Pediatr Clinics North Am. 2010;57: 207-237.
8. Mitchell R, Nivison-Smith I, Anazodo A, et al.
Outcomes of Hematopoietic Stem Cell Transplantation in Primary
Immunodeficiency: A Report from the Australian and New Zealand
children’s Haematology Oncology Group and the Australasian Bone Marrow
Transplant Recipient Registry. Biol Blood Marrow Transplant.
2013;19:338-43.
9. Yadav SP. Bone marrow transplantation for primary immune
deficiency disorders in India: Past, present and future. Indian Pediatr.
2018;55:657-58.


