A 3-year-old boy was admitted
with recurrent febrile illnesses and neurological symptoms. The first
illness, at 21 months of age, manifested as a high-grade fever with
non-paroxysmal cough, respiratory distress, and a generalized macular
rash, present over one week. The perinatal period and development were
normal. Computed tomography scan of the chest showed nodules in the
bilateral lower lobe with necrotic conglomerating mediastinal lymph
nodes suggestive of infective etiology. A clinical diagnosis of probable
measles with pneumonia was considered. At 24 months of age, he again
presented to our center with a febrile illness. Examination showed the
absence of tonsils and peripheral lymph nodes, and presence of a BCG
scar. Family history revealed that two of his maternal uncles had died
in early childhood due to an undiagnosed infection. A clinical diagnosis
of X-linked agammaglobulinemia (XLA) was considered and confirmed by
specific investigations as per the diagnostic criteria proposed by
European Society for Immuno-deficiencies [1]. Monthly intravenous
immunoglobulin (IVIg) (400 mg/kg) was initiated, and he showed
symptomatic improvement. At 26 months of age, he developed left-sided
focal motor status epilepticus and left-sided hemiparesis without
associated fever, altered sensorium, cranial nerve involvement, or
features of raised intracranial pressure. Investigations are shown in
Table I. Magnetic resonance imaging (MRI) of the brain showed focal
signal changes (details in the section on investigations). A clinico-radiological
diagnosis of XLA with probable focal enteroviral encephalitis was
considered. Intravenous acyclovir (30 mg/kg/d for 15 days) and high-dose
IVIg (1g/kg) were given. He recovered with residual left hemiparesis. He
was readmitted a month later with persistent vomiting, intermittent
lethargy, redness of eyes with watery discharge, and brief intercurrent
seizures over the past one week. He had been on regular monthly IVIg
replacements, and trough IgG level was 600 mg/dl. He continued to have
altered sensorium and residual left hemiparesis with radiological
progression of the focal encephalitic changes (details in the section on
investi-gations). He had two further admissions, one and six months
later, with persistent left-sided, focal seizures and residual left
hemiparesis. By 34 months of age, he developed subacute neurological
deterioration (progressive lethargy and reduced interaction), visual
deterioration, and recurrent right-sided focal tonic-clonic seizures
followed by right hemiparesis. At this time, examination showed weight
10 kg (-2 to -3 z), length 90 cm (-1 to -2 z) and head
circumference 48 cm (-1 to -2 z). His Glasgow Coma Scale was 11
(E3M5V3-4),
and pupils were 2mm bilaterally equal and reacting to light. He had
chronic left-sided motor weakness with lower limb spasticity,
acute-onset right-sided weakness with diminished tone, brisk deep tendon
reflexes and bilateral extensor plantar response. Rest of the systemic
examination was unremarkable.
 |
| |
Investigations (Table I): XLA being
a prototype primary humoral immunodeficiency disorder, T cells and their
subsets were not tested in the blood or on the brain sections at
autopsy. Stool sample was negative for both polio and non-polio viruses.
Baseline MRI of the brain at the onset of seizures showed presence of a
right-sided, frontal, subcortical white matter lesion, which showed
gliosis later. New lesions appeared in bilateral occipital and parietal
subcortical white matter, and the thalamus, suggesting a progressive,
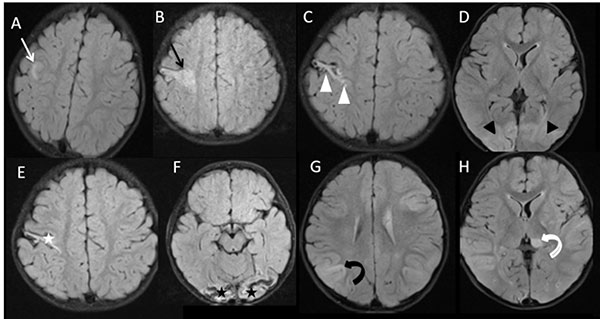
subcortical, multifocal involvement (Fig. 1A-H). MR spectroscopy
revealed reduced NAA with a small lactate peak.
 |
|
Fig. 1 Sequential axial flair MR images
of the patient: Baseline MRI a) shows the presence of
frontal subcortical white matter lesion (white arrow). MRI after
24 days b) shows the progression of frontal subcortical
white matter lesion (black arrow). Interval MRI after one month,
c,d) shows gliosis in the right frontal subcortical
lesion (white arrowheads) and appearance of bilateral occipital
subcortical lesions (black arrowheads). Interval MRI after 1.5
months (e,f) shows gliosis in the frontal (white star) and
occipital (black stars) subcortical lesions. Interval MRI after
five months (g,h) shows the appearance of new parietal
subcortical (curved black arrow) and thalamus (curved white
arrow) lesions. No evidence of any diffusion-weighted
abnormality, susceptibility-weighted abnorma-lity,
contrast enhancement, or angiographic abnormality was noted in
all the MRI examinations.
|
Course and Management: Multiple
anti-epileptic drugs (phenytoin, valproate, levetiracetam,
phenobarbitone, midazolam infusion) were sequentially given for
right-sided epilepsia partialis continua. He was intubated and
ventilated for worsening sensorium. Intravenous acyclovir and high dose
IVIg (2g/kg total dose) were restarted. Brain biopsy was deferred due to
the poor general condition of the patient. Newer therapies such as
pleconaril and pocapavir were considered but could not be tried due to
resource-constraints. The patient further developed high-grade fever
secondary to nosocomial infection and/or aspiration pneumonia.
Intravenous antimicrobials were sequentially upgraded (ceftriaxone,
meropenem, vancomycin). He suffered a cardiac arrest on day 19 of
hospital stay and could not be revived.
Unit’s Final Diagnosis
X-linked agammaglobulinemia with recurrent seizures
and encephalopathy probably due to chronic enteroviral encephalitis
complicated with nosocomial pneumonia.
DISCUSSION
Clinical discussant: We had a 34-month-old boy
with recurrent sinopulmonary infections, absent tonsils, lymph nodes and
peripheral B-cells, pan hypogamma-globulinemia, reduced Btk protein
expression on CD14+ monocytes and progressive neurocognitive decline.
The family history supported an X-linked inheritance pattern. Based on
the standard diagnostic criteria, a diagnosis of underlying XLA is
beyond doubt [1].
Regarding the central nervous system (CNS)
mani-festations, the patient had an acute CNS event, which progressed
both clinically and radiologically over the subsequent 12-14 months with
intermittent exacer-bations. CNS manifestations in children with XLA can
be either due to an inability to clear the opportunistic infections, or
due to a dysregulated immunity, or both. XLA is a prototype of humoral
immunodeficiency disorders with B-lymphocyte differentiation arrest,
resulting in recurrent infections with encapsulated bacteria like
pneumococcus and H. influenza, gastrointestinal infections with
giardia and chronic enteroviral infections [2]. However, the chronic and
indolent clinical course, presence of significant neutropenia and
absence of fever during all episodes of clinical deterioration make
bacterial infection unlikely. Moreover, the patient being on regular
IVIg replacement therapy had adequate trough level (>600 mg/dL) to
control the bacterial infections [3]. Although there are anecdotal
reports of fungal and mycobacterial infections causing primary CNS
manifestations in XLA patients, overall, these infections primarily
concern the cell-mediated immunity, which is intact in XLA patients.
In the index case, toxoplasma serology (IgG and IgM)
and PCR were negative. In neuroimaging toxoplasmosis lesions usually
appear as hypointense (on T1- weighted) with high or mixed signal
intensity (on T2-weighted and FLAIR images) signals, typically in basal
ganglia, cortico-medullary junction, white matter, and the
periventricular regions [4]. Few case reports of toxoplasmosis in
acquired immunodeficiency syndromes have shown hyperintense lesions
involving basal ganglia, thalamus and cerebral hemispheres [5,6].
However, in our case, neuroimaging was not in favour of toxoplasmosis.
Enteroviral infections are known to cause difficult,
persistent CNS disease in children with XLA. As opposed to other
viruses, which are dealt with by cell-mediated immunity, the host
response to enteroviral infections in XLA is by forming neutralizing
antibodies.
In one of the
largest series of 36 patients of XLA seen over two decades, CNS
infections constituted a significant proportion (25%) of all infections
[7] with enteroviral infections including echo, polio and coxsackie
being the most problematic [8]. As seen in our case, enteroviral encephalitis in XLA are described as
insidious onset, slowly progressive loss of motor and cognitive
mile-stones over 2-3 years, followed by spastic quadriplegia, coma with
mortality in nearly 44% of cases [9, 10]. CSF samples remained negative
for all enteroviruses, tested both by PCR and by viral cultures in the
index case, which may be falsely negative and does not exclude the
infection. Though adequate trough levels by IVIg replacement therapy may
prevent bacterial infections it protect against enteroviral infections
[3].
Besides enteroviruses, astrovirus, measles and herpes
viruses need to be considered in XLA [11, 12]. Though, the index case
had a measles-like past illness, and had been immunized with a live
vaccine, measles inclusion body encephalitis was unlikely, because it is
a disease of patients with depressed cell-mediated immunity with a rapid
and fatal course. The clinical and radiological presentation in the
index case did not favor subacute sclerosing pan-encephalitis. Moreover,
the measles serology was negative and measles virus inclusion bodies
were absent on the histopathology. Other than these, clinical
presentations described in the anecdotal reports of other viruses such
as adenovirus, influenza virus, cytomegalovirus, and John Cunningham
(JC) virus [13] did not fit into our clinical scenario. Progressive
multifocal encephalopathy, reported with XLA was also unlikely [14,15].
Dysregulated immunity leading to autoimmune encephalitis, abnormal
immune response to drugs [2] or IVIg therapy induced progressive,
chronic neuro-regression [3] were other differential diagnosis, but
unlikely in this case.
Pediatric immunologist 1: In the present case,
the trough level of immunoglobulins being well above 600 mg/dL, he was
protected against the common bacterial pathogens, thus making bacterial
infection of CNS unlikely. The most common CNS pathology in such
patients is an enteroviral infection, which can occur even when the
patient is on regular IVIg therapy. Therefore, this is consistent with a
case of chronic enteroviral infection with XLA.
Neurologist 1: The diagnosis of chronic
enteroviral infection with XLA seems most likely in this case. The early
onset of gliosis in the MRI brain of this patient suggests a vascular
invasion. As pointed rightly by the clinical discussant, measles
inclusion body encephalitis is a fulminant infection that does not
follow such a chronic indolent course. Additionally, there is no
contrast enhancement in any of five sets of MRIs probably due to the
lack of immunity to mount an inflammation. JC virus could be another
possibility.
Virologist: Enterovirus is the most common
etiology for this clinical presentation. Astrovirus, as the discussant
highlighted, is also being reported. Sensitivity of detection of
enterovirus by CSF PCR ranges from 75-100%. It would be ideal to take a
throat swab along with CSF samples. Studies have shown that sensitivity
has improved when throat swab is being examined along with CSF PCR.
Pediatric pulmonologist: Although the case
strongly points towards a viral infection, yet the presence of necrotic
lymph nodes in computed tomogrpahy chest and persistent mastoiditis
might be suggestive of other infectious agents such as fungus or an
invasive hospital-acquired infection. As the autopsy was done for brain
only, infection elsewhere in the body could not be identified.
Pediatric hemato-oncologist 1: One could consider
granulomatous amoebic infections such as Balamuthia, which have
been previously reported from the center, although the MRI picture does
not conform to it.
Pediatric immunologist 2: The patient received
4-5 doses of oral polio vaccine in routine immunization schedule, along
with a dose on the national immunization day, 15 days prior to the onset
of illness. Live viral vaccines are contraindicated in such patients as
well as in siblings and their surroundings.
Physician: Measles seems more likely in
the index case than enteroviruses as the posterior parts of the brain
are more involved. The absence of enhancement and cells in the CSF and
the presence of high protein and gliosis in the brain are described in
measles. However, the clinical course in measles is shorter for 1-3
months, as compared to the chronic course in the index lasting nearly 12
months.
Clinical discussant: Measles inclusion body
ence-phalitis is primarily a disease of cell-mediated immunity, seen
more commonly in adult patients, and follow a rapid fulminant course.
Subacute-chronic measles virus infection could not be completely
excluded. Serological testing for measles virus was not feasible as
B-cells are deficient in XLA. JC virus infection would be unusual as the
virus is carried to the brain by B-cells, which are deficient in XLA
patients. Polioviruses are also a type of enteroviral infections. XLA
patients are prone for atypical manifestation of enteroviruses, which
includes fulminant polio encephalitis as well as paralytic
poliomyelitis. The abnormal chest findings on computed tomography
described the lung pathology at the time of first illness when the child
was admitted with a viral prodrome, bacterial pneumonia and neutropenia
suggestive of an acute bacterial necrotizing pneumonia. Additionally,
investigations for tuberculosis and fungal infections had been
non-corroborative at that time. It would be very unusual for a
mycobacterial or fungal infection to present with such CNS
manifestations over several months. However, systemic infection with
pneumocystis carinii is described in patients with XLA.
Pediatric hemato-oncologist 2: As progressive
multifocal leukoencephalopathy secondary to JC virus infection is common
in hematological conditions treated with rituximab, a similar mechanism
may be proposed for XLA patients also. The patient also had persistent
microcytic, hypochromic anemia with thrombocytopenia, which could be due
to an enteroviral inflammatory bowel disease or an intestinal giardiasis,
which is common in XLA patients.
Pediatric immunologist 1: As part of an
international collaborative study, 32 children of the institute, with
XLA were screened for poliovirus. None of them had poliovirus
infections. In Iran, 4% of children with XLA had poliovirus infection
[16]. The negative stool samples for poliovirus make this infection
unlikely in the case.
Pediatric neurologist 1: Chronic herpes
encephalitis type 1 and human herpes virus-6 infection may be additional
possibilities.
PATHOLOGY PROTOCOL
A partial autopsy was performed in this case. The
external examination of the brain weighing 1142 grams, showed slightly
congested meninges, without any exudate. A mild tonsillar herniation was
noted. Blood vessels of circle of Willis and brainstem appeared normal.
Bilateral parieto-occipital and temporal lobes were discolored,
collapsed and soft (Fig.2). The coronal sectioning of the
brain revealed softening, shrinkage and thinning of the cortical ribbons
of left inferior frontal, left frontal, paramedian area above cingulate
gyrus and right middle and inferior frontal gyrus (Fig. 2). Both
temporal lobes and bilateral parietal cortices had similar changes with
a shrunken left temporal lobe. The right occipital lobe showed cystic
encephalomalacia. While the left putamen and adjacent internal capsule
showed necrosis explaining his right hemiparesis, the right lentiform
nucleus was normal (Fig. 2). The affected areas of the brain
corresponded to the anterior, middle, and posterior cerebral artery
territories indicating global hypoxia. The hippocampi, thalami and
midbrain appeared normal grossly. The white matter was mostly spared.
The brainstem axial cuts revealed mild congestion of the dorsal parts of
the pons, with unaffected medulla and cerebellum.
 |
|
Fig. 2 a) The parietal convexity
is discolored and collapsed due to softening of underlying
cortex of the brain; b) Coronal section of the brain
shows shrinkage and softening of the cortical ribbons affecting
left inferior frontal gyrus and both temporal lobes. The white
matter is relative spared, c) The left putamen and
internal capsule are soft and necrotic in this coronal slice.
The right basal ganglia and both thalami are spared. Note that
left temporal is affected more than right temporal lobe; d)
Sparse lymphocytic inflammation is seen in the cortical
leptomeninges; e) The affected cortical areas showed
neuronal loss and necrosis and replacement by macrophages and
lymphocytes; f) Reactive hypertrophied astro-cytes. The
occipital cortex demonstrated foci of micro-calcification and
microglial proliferation, g-h) The dentate nucleus of the
cerebellum shows neuronal loss, microglial proliferation, and
vascular congestion.
|
Microscopic examination revealed sparse meningeal
infiltrates, predominantly lymphocytic (Fig. 2). The grossly
affected cortical areas showed laminar and transcortical necrosis,
replaced by large number of foamy macrophages admixed with few
lymphocytes, accompanied by reactive glial proliferation (Fig. 2).
The adjacent cortical areas showed evidence of hypoxic changes, more
marked at the base of the sulci than the crests. Hippocampi showed
diffuse hypoxic damage and patchy neuronal loss. The posterior parts of
the occipital cortex showed extensive neuronal loss, cyst formation and
calcium deposits indicating chronicity (Fig. 2). Therefore, the
cerebral cortex, in nutshell, showed presence of hypoxic damage and
varying degree of cortical necrosis, explained by recurrent seizures.
Histological examination of the dorsal pons demonstrated neuronal loss,
neuronophagia and microglial proliferation with nodule formation,
highlighted by CD68 immuno-staining. The perivascular lymphocytic
infiltration consisted of CD3-positive T-cells without any CD20-positive
B-cells. Similar changes were noted in the midbrain, dentate nucleus of
the cerebellum and grey matter around 4
th
ventricle with cerebellar folia being unremarkable. The dorsal motor
root of vagal nucleus and the anterior horn cells of the cervical
segment of the cord were affected. Immunohistochemistry for herpes
simplex virus 1 and 2, cytomegalovirus, simian virus 40 (SV40), Epstein
Barr and parvovirus were negative. In addition, PCR for enteroviruses
was negative in the brain tissue. The post-mortem biopsy samples of
brain, lung and liver tissues did not contribute any significant
information.
The topography of the lesions in this brain namely
the involvement of dorsal pons, dentate nucleus, medulla, part of the
hypothalamus and sparing of thalamus and cortical zone favours
enteroviral infection, even though PCR was negative. PCR positivity
depends on multiple factors and at best gives 50% positive results. The
lesions and the histological features favour a chronic enteroviral
infection over JC virus. SV40 antibody, which recognizes both JC virus
and BK polyoma virus failed to show any positivity. The final autopsy
diagnosis was XLA with probable enteroviral encephalitis and cystic
cerebral encephalomalacia.
Open Forum
Pediatric neurologist 2:
Considering a remarkably similar case of XLA with seizures reported by
the CDC, where RNA separation method detected astrovirus, infection with
other single-stranded RNA viruses could be a possibility. The presence
of hemorrhage and calcification in the occipital lobe could also suggest
Posterior reversible encephalopathy syndrome-like changes.
Neurologist 2: As none of the viral studies
yielded an etiological agent, autoimmune encephalitis could be a
possibility. The response to ongoing IVIg therapy could also be a
pointer toward an underlying autoimmune process.
Pathology discussant: The case has been a
clinical and histopathological challenge. While Posterior reversible
encephalopathy syndrome mostly involves the white matter, gray matter
involvement was seen in this case. Autoimmune encephalitis should only
be considered when infectious causes have been excluded. A negative PCR
does not exclude an enteroviral infection. Enterovirus A71 infection has
been prevalent in India, Bangladesh, Malaysia, and Taiwan. Enterovirus
D68 can present similarly in an XLA patient. Other uncommon
enteroviruses and single-stranded RNA viruses also remain a possibility.
In the presence of normal limbic organs, cingulate gyrus, and amygdala
on histopathology, a limbic encephalitis is most unusual. Other forms of
autoimmune encephalitis such as anti-NMDAR and anti-AMPAR need specific
testing. The California encephalitis project has reported that 63% of
the probable encephalitis cases remain unknown despite extensive
investigations for infectious causes. The hypoxic changes in the brain
in the case were probably due to recurrent seizures.
Pediatric immunologist 3: The histopathology
shows typical features of viral encephalitis with infiltration by CD3
lymphocytes alone, CD20 lymphocytes being absent, as expected in XLA
patients. The diagnosis of astrovirus infection in the CDC case alluded
to in the previous discussion was based on highly advanced pyro
sequencing PCR technique which is not routinely available.
DISCUSSION
The case highlights the unique presentation of a
child with XLA and recurrent infections. A simple throat examination for
the presence of tonsils is a vital bedside clue and helps clinch the
diagnosis. CNS infections are tough to treat and constitute a
significant cause of mortality in these children as seen in the index
case [17]. Although the presence of good T-cell functions protects the
patients from common childhood viral infections, yet enteroviruses
notoriously cause chronic infections [18,19]. The brain pathology was
not consistent with the diagnosis of JC virus-related multifocal
leukoence-phalopathy, where multifocal discrete white matter
demyelination occurs initially, progressing to form confluent large
demyelinating lesions appearing as granular soft discolored plaques. As
mentioned in the pathology description, the white matter was spared in
this case with a predominantly grey matter disease. No demyelination is
seen in the index case. There were no oligodendroglia inclusions or
bizarre astrocytes and anti-SV40 antibody on immunohistochemistry did
not show any viral antigen in brain tissue. The typical involvement of
brainstem nuclei, dentate nucleus of cerebellum and hypothalamus showing
neuronal loss, microglial hyperplasia is characteristic of an
enteroviral infection [19]. Although JC virus could not be tested in the
CSF, SV40 antibody used for immunostaining on the brain sections did not
show any positivity for the same. The collections of foamy macrophages
with a few lymphocytes on histopathology are from multiple infarcts
involving various regions of the cerebral cortex. This is the second
pathology in brain, which occurred due to severe hypoxia in this child
because of repeated seizures. The cerebral infarcts were of different
durations.
Hence, with the available investigative work-up
possible in a resource-limited setting, we could conclude that the case
was a probable enteroviral meningo-encephalitis. The topography of the
lesions, the peculiar preponderance of enteroviral infections in
children with XLA, the histological features and immunohisto-chemistry
favour an enterovirus over JC virus, although the virus could not be
demonstrated by PCR in the brain tissue. This is a common scenario in
several pediatric centers in India and needs to be brought out, even if
an organism could not be identified. Prevalence of enteroviral
encephalitis in XLA is reported between 1% and 3% [8]. Echovirus,
poliovirus, coxsackievirus and several uncommon enteroviruses may cause
chronic progressive encephalitis with neuroregression [17,20] with
enteroviruses being one of the most common causes of meningoencephalitis
in patients with XLA [21]. The sensitivity of CSF PCR-based assays for
enteroviruses ranges from 75%-80%. Combining the CSF PCR with a throat
swab may increase the sensitivity of detection [22, 23]. Regular IVIg
therapy with adequate trough levels protects against severe bacterial
infections [3]. However, the role of high dose peripheral and
intraventricular immunoglobulin for enteroviral encephalitis is
debatable [9,17].
Contributors: AGS: Concept and design of the
study, data collection and interpretation, drafting manuscript data
interpretation, editing of draft, critical revision, Clinical discussant
of CPC; BDR: Design of the study, drafting manuscript, acquisition of
data and data analysis; pathology discussant of CPC; DB: Concept and
design of the study, patient care, data collection and interpretation,
drafting manuscript data interpretation, editing of draft, critical
revision; AR: Acquisition of immunological data and data analysis,
critical revision of manuscript; VB: Acquisition of radiological data
and data analysis, critical revision of manuscript, radiology discussant
of the CPC.
REFERENCES
1. Conley ME, Notarangelo LD, Etzioni A.
Diagnostic Criteria for Primary Immunodeficiencies. Representing PAGID
(Pan-American Group for Immunodeficiency) and ESID (European Society for
Immunodeficiencies). Clin Immunol. 1999;93:190-7.
2. Hernandez-Trujillo VP, Scalchunes C,
Cunningham-Rundles C, et al. Autoimmunity and inflammation in X-linked
agammaglobulinemia. J Clin Immunol. 2014;34:627-32.
3. Suri D, Bhattad S, Sharma A, et al. Serial serum
immunoglobulin G (IgG) Trough levels in patients with X-linked
agammaglobulinemia on replacement therapy with intravenous
immunoglobulin: Its correlation with infec-tions in Indian children. J
Clin Immunol. 2017;37:311-8.
4. Osborn AG, Jhaveri MD, Salzman KL, editors.
Diagnostic imaging. Brain. Third edition. Elsevier; 2016.
5. Hofmann A, Zaharatos G, Miller M. Case report and
review of the literature: Toxoplasma gondii encephalitis in a
40-Year-Old woman with common variable immuno-deficiency and a new
diagnosis of large granular lympho-cytic leukemia. Can J Infect Dis Med
Microbiol. 2008;19: 309-10.
6. Holtkamp M, Okuducu AF, Klingebiel R, Ploner CJ.
Cerebral toxoplasmosis in a patient with common variable
immunodeficiency. Neurology. 2004;63:2192-3.
7. Singh S, Rawat A, Suri D, et al. X-linked
agamma-globulinemia: Twenty years of single-center experience from North
West India. Ann Allergy Asthma Immunol. 2016;117:405-11.
8. Winkelstein JA, Marino MC, Lederman HM, et al.
X-linked agammaglobulinemia: Report on a United States registry of 201
patients. Medicine (Baltimore). 2006;85: 193-202.
9. Bearden D, Collett M, Quan PL, Costa-Carvalho BT,
Sullivan KE. Enteroviruses in X-linked agamma-globulinemia: Update on
epidemiology and therapy. J Allergy Clin Immunol Pract. 2016;4:1059-65.
10. Halliday E, Winkelstein J, Webster ADB.
Enteroviral infections in primary immunodeficiency (PID): A survey of
morbidity and mortality. J Infect. 2003;46:1-8.
11. Conley ME, Dobbs AK, Farmer DM, et al. Primary B
cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol.
2009;27:199-227.
12. Dropulic LK, Cohen JI. Severe viral infections
and primary immunodeficiencies. Clin Infect Dis. 2011;53:897-909.
13. Chun J-K, Lee TJ, Song JW, Linton JA, Kim DS.
Analysis of clinical presentations of Bruton disease: A review of 20
years of accumulated data from pediatric patients at Severance Hospital.
Yonsei Med J. 2008;49:28-36.
14. Teramoto T, Kaneko H, Funato M, et al.
Progressive multifocal leukoencephalopathy in a patient with X-linked
agammaglobulinemia. Scand J Infect Dis. 2003;35:909-10.
15. Zerbe CS, Marciano BE, Katial RK, et al.
Progressive multifocal leukoencephalopathy in primary immune
deficiencies: Stat1 Gain of function and review of the literature. Clin
Infect Dis. 2016;62:986-94.
16. Mamishi S, Shahmahmoudi S, Tabatabaie H,
Teimourian S, Pourakbari B, Gheisari Y, Yeganeh M, Salavati A,
Esteghamati AR, Gooya MM, Nategh R, Parvaneh N. Novel BTK mutation
presenting with vaccine-associated paralytic poliomyelitis. Eur J
Pediatr. 2008;167(11):1335-8
17. Sag AT, Saka E, Ozgur TT, et al. Progressive
neurode-generative syndrome in a patient with X-linked
agamma-globulinemia receiving intravenous immunoglobulin therapy. Cogn
Behav Neurol. 2014;27:155-9.
18. Suri D, Rawat A, Singh S. X-linked
agammaglobulinemia. Indian J Pediatr. 2016;83:331-7.
19. Sanna PP, Burton DR. Role of antibodies in
controlling viral disease: Lessons from experiments of nature and gene
knockouts. J Virol. 2000;74:9813-7.
20. Wong KT, Ng KY, Ong KC, et al. Enterovirus 71
encephalomyelitis and Japanese encephalitis can be distinguished by
topographic distribution of inflammation and specific intraneuronal
detection of viral antigen and RNA. Neuropathol Appl Neurobiol.
2012;38:443-53.
21. Misbah SA, Spickett GP, Ryba PC, et al. Chronic
enteroviral meningoencephalitis in agammaglobulinemia: Case report and
literature review. J Clin Immunol. 1992;12:266-70.
22. Jain S, Patel B, Bhatt GC. Enteroviral
encephalitis in children: clinical features, pathophysiology, and
treatment advances. Pathog Glob Health. 2014;108:216-22.
23. Plebani A, Soresina A, Rondelli R, et al. Clinical,
immunological, and molecular analysis in a large cohort of patients with
X-linked agammaglobulinemia: An Italian multicenter study. Clin Immunol.
2002;104:221-30.


