C
hildhood interstitial lung disease (ChILD) is
clinically recognized when there is prolonged history of a
non-productive cough, tachypnea more than chest wall retractions,
minimal to absent chest auscultatory signs, normal cardiac examination,
the presence of clubbing and hypoxemia responsive to oxygen [1].
Etiologies of ChILD are diverse, and corroborative clinical or
laboratory features help in guiding focused investigations [2]. We
describe the clinicopathological case conference of an infant with
interstitial lung disease (ILD), where the various etiologies of ChILD
and importance of lung histopathology are discussed.
Clinical Protocol
History: A one-year-old boy was hospitalized with
complaints of fever, cough, difficulty in breathing of one month
duration, and intermittent bluish discoloration of lips, palms, and
soles, especially on crying. Fever was intermittent, documented up to
104°F, responding to antipyretics. Cough was non-paroxysmal, associated
with difficulty in breathing and feeding disturbance. The child had been
previously admitted twice for cough and breathing difficulty; first
episode at 5 months of age, and another at 9 months of age. He was
treated with intravenous (IV) antibiotics and oxygen; however, the
breathing difficulty persisted. He was born by a full term normal
delivery at a hospital with a birthweight of 2.5 kg and had normal
neonatal transition. He had been exclusively breastfed until 7 months of
age and was on a milk-based diet along with breastfeeds thereafter. He
was the only child of the non-consanguineous parents. Father was a
smoker and there was no history of contact with pulmonary tuberculosis
(TB). The child had been immunized for age, but BCG scar was absent.
Examination: Anthropometric evaluation revealed
the weight for age <3rd centile, length for age was at 5th centile,
weight for length was 72% of expected, which indicated acute-on-chronic
malnutrition. The occipitofrontal circumference was at 5th centile. At
admission, heart rate was 160/min, respiratory rate was 80/min, blood
pressure was 90/60 mm Hg, with good normal volume pulses, and normal
capillary refill time. Oxygen saturation (SpO
2)
was 50% on room air, which improved to 97% on oxygen support using nasal
prongs (5L/ min). Grade III pan-digital clubbing and anemia were noted.
Respiratory system examination showed minimal subcostal retractions,
without any crepitations or wheeze on auscultation. Abdominal,
cardiovascular and central nervous system examinations were normal.
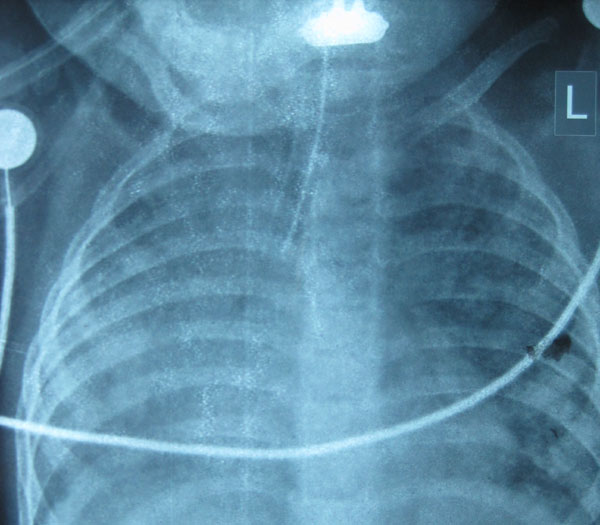 |
|
Fig. 1 X-ray anterio-posterior view
of chest showing bilateral interstitial infiltrates and ground
glass opacities.
|
Investigations: Investigations revealed
hemoglobin of 8.4 g/dL which dropped significantly to 6.4 g/dL during
the course of hospital stay, requiring two packed red cell transfusions.
There was persistent polymorphonuclear leukocytosis (absolute neutrophil
count: 16,000-18,500/mm3)
and thrombocytosis (7.51 lakhs/mm3).
Peripheral smear revealed microcytic hypochromic anemia. Alanine
transaminase and aspartate transaminase levels were 24 U/L and 34 U/L,
respectively. Arterial blood gases showed persistent respiratory
acidosis. Chest radiograph showed bilateral haziness with infiltrates (Fig.
1). High-resolution computerized tomography (HRCT) of the chest
revealed features of bilateral diffuse ILD, pulmonary artery
hypertension and mediastinal lymphadenopathy. Echocardiogram showed
small atrial septal defect with a left to right shunt. Work-up for the
causes of ChILD is summarized in Table I.
TABLE I Investigations for Diffuse Interstitial Lung Disease in the Index Child
|
Infectious diseases work-up |
|
*AFB staining, fungal smears and P. jirovecii:
Negative - repeated thrice |
|
|
|
*AFB cultures and fungal cultures: Negative |
|
Tuberculin skin testing 0 mm |
|
HIV, CMV serologies: Negative |
|
Immunology work-up |
|
Absolute lymphocyte counts 4,600/mm3 |
|
IgG, IgA, IgM values: 692 mg%, 76 mg%, 193 mg%, respectively |
|
Connective tissue disorders/Pulmonary hemosiderosis work-up |
|
Urine examination: Albumin- negative, no RBC casts |
|
*Hemosiderin-laden macrophages: Negative |
|
ANA, ANCA: Negative |
|
Other work-up |
|
Sweat chloride levels: 40 mEq/L |
|
Ultrasound abdomen: Normal study |
|
Fundus examination: No cherry red spot |
|
AFB: Acid-fast bacilli; HIV: Human Immunodeficiency Virus;
CMV: Cytomegalovirus; Ig: Immunoglobulin; RBCs: Red blood cells;
ANA: Antinuclear antibody; ANCA: Anti-neutrophil cytoplasmic
antibody; *Gastroic lavage specimens. |
Course and management: Child had a hospital stay
of around 2.5 months. At admission, he was started on nasal prongs
oxygen (5 L/min) and IV ceftriaxone, amikacin, and cloxacillin. Later
azithromycin was added to cover atypical pneumonia and cotrimoxazole for
suspected Pneumocystis jirovecii pneumonia (PJP). As the clinical
and radiological features suggested an ILD, oral prednisolone was
started at a dose of 2 mg/kg for 14 days followed by tapering over next
4 weeks. The infant responded to steroids initially as his respiratory
rate and breathing difficulty decreased though the oxygen requirement
persisted. However, on the day 20 of steroid tapering, the child
developed marked respiratory distress, fever, lethargy, increasing CRP
and features of disseminated intravascular coagulation (DIC) with
platelet count of 27,400/mm3.
The child was subsequently intubated and given supportive care in the
form of platelets and albumin transfusion. The dose of steroids was
again hiked up and antibiotics were added. The child also had
pre-terminal worsening of liver and renal functions. The endotracheal
aspirate culture showed Acinetobacter spp. Child persisted to
have severe sepsis with refractory shock and suffered cardiac arrest and
could not be revived.
Unit’s final diagnosis: The unit’s final
diagnosis was ChILD with healthcare-associated infection – sepsis with
multiorgan dysfunction syndrome (MODS). The cause of death was kept as
acute respiratory distress syndrome (ARDS).
Clinical discussion: We have an
infant with features of ILD - prolonged history of cough and breathing
difficulty, failure to thrive (FTT), tachypnea, hypoxemia which improved
with oxygen, pan-digital clubbing, normal cardiac examination,
supportive chest radiograph and HRCT features. Child’s respiratory
status improved with corticosteroids. Another important point to note is
that this child had anemia which required packed red blood cell
transfusion twice. This case could be approached under two subheadings –
etiological approach and the preterminal events.
Index child had an insidious onset diffuse ILD, which
encompasses a broad group of diseases that affect the respiratory
function of the lung by impairing alveolar gas exchange [2]. As the
child was one-year-old, causes like developmental disorders, disorders
which are related to systemic diseases, diseases of immunocompromised
host, diseases of normal hosts, aspiration syndromes and infections need
to be discussed (Table II). ChILD differs from the adult
forms of ILDs because here the injury occurs during the development and
differentiation of lung and etiology also includes various genetic and
metabolic causes [3]. Disorders of lung growth and development are
unlikely in our child as the neonatal course was uneventful.
Neuroendocrine cell hyperplasia of infancy and pulmonary interstitial
glycogenosis also seems unlikely as the presentation would be much
earlier in these disorders. Genetic disorders of surfactant production
and metabolism could be a possibility; however, surfactant protein B
mutations are unlikely as the presentation in those cases would be in
early infancy. Whereas pulmonary microlithiasis, vascular, lymphatic
disorders, pulmonary infiltrates with eosinophilia could be ruled out
because supportive laboratory or radiological evidence were not seen.
Human immunodeficiency virus (HIV) and tuberculosis (TB) are important
infectious causes that can present with failure to thrive (FTT), ChILD
and mediastinal adenopathy [4,5]. Lymphoid interstitial pneumonitis is a
known feature of HIV. However, HIV serology was negative and multiple
gastric lavage samples for acid-fast bacilli staining were not
suggestive. TB would still be a strong possibility as it is endemic in
India and pediatric TB is usually paucibacillary [6]. Cytomegalovirus
(CMV) infection, either acquired by perinatal transmission or in a
setting of primary immunodeficiency (PID), could result in persistent
interstitial pneumonia. However, there was no accompanying
thrombocytopenia or transaminitis, and CMV serology was negative. PJP
could also result in interstitial shadows, but symptoms are usually
acute and it occurs in a setting of primary or secondary cellular PID
[2]. Absolute lymphocyte counts of the child and immunoglobulin profile
were normal, thereby ruling out most of the common PIDs.
TABLE II Etiologies for Interstitial and Diffuse Lung Diseases of Infants
|
Diseases common in the infancy age group |
|
Developmental disorders
|
|
Congenital alveolar dysplasia
|
|
Acinar dysplasia
|
|
Alveolar-capillary dysplasia with pulmonary vein misalignment
|
|
Growth abnormalities |
|
Neonatal lung diseases- Bronchopulmonary dysplasia (in preterm)
and chronic lung disease (term neonates)
|
|
Structural lung changes- Trisomy 21
|
|
Pulmonary hypoplasia
|
|
Lung disease associated with congenital heart disease
|
|
Conditions of undefined etiology |
|
Neuroendocrine cell hyperplasia of infancy
|
|
Pulmonary interstitial glycogenosis
|
|
Surfactant metabolism disorders |
|
Mutations in SPFTB, SPFTC, ABCA3
|
|
Diseases that are not specific to infancy age group |
|
Normal host |
|
Infections and post-infectious
|
|
Hypersensitivity pneumonia
|
|
Aspiration syndromes
|
|
Eosinophilic pneumonia
|
|
Immunocompromised host |
|
Opportunistic infections (HIV, primary immunodeficiency)
|
|
Transplant rejection syndromes
|
|
Systemic diseases |
|
Storage disorders- familial lysinuric protein intolerance,
Niemann Pick disease
|
|
Autoimmune conditions- SLE, JDMS, ANCA-associated vasculitides,
sarcoidosis
|
|
Langerhans cell histiocytosis
|
|
Diseases mimicking ILD |
|
Hypertensive vasculopathy |
|
Lymphatic malformations |
|
Congestive vasculopathy due to cardiac dysfunction or veno-occlusive
diseases |
|
Unclassified |
|
End stage disease, lung biopsies that are inadequate or
non-diagnostic |
|
JDMS: Juvenile dermatomyositis; SLE: Systemic lupus
erythematosus; ANCA: Anti-neutrophil cytoplasmic antibody; HIV:
Human immunodeficiency virus. |
Cystic fibrosis (CF) is another possibility that
could be considered as there is a chronic respiratory illness,
malnutrition, FTT and clubbing. However, odd points are normal sweat
chloride levels, absence of typical imaging features of chest wall
hyperinflation, and absence of pancreatic insufficiency. Pulmonary
hemosiderosis could be a likely cause as the child also had microcytic
hypochromic anemia requiring packed red cell transfusions. The classic
triad consists of iron deficiency anemia, alveolar infiltrates on chest
radiograph and hemoptysis which may not be present in all children.
However, gastric aspirates for hemosiderin-laden macrophages (HLM) done
thrice were negative. Adult type idiopathic ILD can also occur in
children and this possibility could not be excluded. Ground glass
opacities with sparing of the upper and middle lobes, and cystic changes
in lungs, as seen on HRCT chest of the index child, could be seen in
non-specific interstitial pneumonitis and lymphocytic interstitial
pneumonia. Drug-induced lung diseases, sarcoidosis, and hypersensitivity
pneumonitis are unlikely because they are more common in adult age
group.
Systemic disorders that could lead to ILD are less
likely in the index child as there were predominant lung complaints and
there was no clinical evidence of other organ system involvement.
Langerhans cell histiocytosis (LCH) of the lung may have the clinical
presentation of an ILD; however, HRCT usually shows multiple cystic
spaces [2]. Index child had predominant ground glass opacities and there
was no ear discharge, seborrhea or hepatosplenomegaly. Familial
lysinuric protein intolerance is a rare inborn error of metabolism that
can result in progressive interstitial pneumonitis; however, there were
no other suggestive features like hepatosplenomegaly, sparse brittle
hair, or centripetal obesity. Niemann-Pick disease could also have an
interstitial lung involvement; however, there is no hepatosplenomegaly
in our child [2]. Neurocutaneous syndromes are unlikely because there
were no neurocutaneous markers.
Pre-terminally the child had nosocomial sepsis, a
tracheal aspirate culture that showed Acinetobacter spp, and
shock which was multifactorial (hypoxic, septic and neurogenic). The
child also had MODS in the form of acute renal failure (ARF), ARDS and
encephalopathy, which ultimately led to brain death. I would like to
conclude by saying that it is a ChILD most likely due to: (i)
infectious etiology – either TB or CMV and (ii) CF or an
idiopathic pulmonary hemosiderosis that cannot be ruled out with
associated pulmonary artery hypertension, nosocomial sepsis, and MODS.
Open Forum
Pediatric pulmonologist: Chest X-ray of
the child showed diffuse alveolar opacities, with alveoli difficult to
differentiate from the interstitium. TB is one disorder that can present
with ILD, but none of the investigations were positive. In fact, the
child showed a good response to steroids with improvement in oxygen
requirement but succumbed to hospital-acquired sepsis. As already
discussed, there are so many features not supporting the diagnosis of CF
in this child.
Pediatrician 1: I would like to include primary
ciliary dyskinesia as a differential diagnosis here as there are
features of early cystic dilation of alveoli, FTT, and recurrent
pneumonia. Pulmonary hemosiderosis that can have recurrent
pneumonia-like presentation associated with a recurrent drop in
hemoglobin is another possibility. Actually, ILD is a broad bag
consisting of a variety of disorders hence we really cannot pinpoint
from clinical presentation alone.
Pediatric hemato-oncologist: This child did have
some interstitial cystic spaces and mediastinal lymph nodes. Hence, the
possibility of LCH cannot be still ruled out. Skeletal survey if done
could have identified classical punched out lesions.
Pathology Protocol
A complete autopsy was performed on this one-year-old
child. The brain was essentially normal on gross and microscopic
examination. There was 300 mL of serous fluid in both pleural cavities.
No excess of fluid was seen in pericardial and peritoneal cavities.
Both the lungs weighed 315 gms, were heavy, firm with
patchy pleuritis. There was cobblestone appearance visualized more on
the right side. On slicing, the lungs were non-crepitant with a solid
appearance. Upper lobes showed microcystic change with honeycombing and
intervening scarred areas. Microscopically there was a marked
proliferation of type 2 pneumocytes with focal muscularization of
alveolar septae. Few foci showed lymphomononuclear infiltrate within
interstitium and giant cell transformation (Web Fig.
1). No viral inclusions were seen. An occasional focus of osteoid
formation was noted. Other areas revealed fresh and old alveolar
hemorrhages with septal fibrosis. Features of pulmonary artery
hypertension were present in the form of medial hypertrophy and intimal
proliferation. Patchy early bronchopneumonia and pulmonary
thromoboem-bolism was also seen possibly as a result of terminal events
(Web Fig. 1).
The heart weighed 78 g and was normal in shape. All
the valves were within normal limits, no vegetations were seen. The
right ventricle wall thickness measured 0.4 cm, suggesting hypertrophy.
The liver was 350 g in weight, pale and greasy to feel with exaggerated
mottling on the cut surface. Microscopically there was diffuse
microvesicular steatosis with sinusoidal dilatation and congestion.
Spleen weighed 20 g with small subcapsular hemorrhages. Kidneys weighed
80 g, appeared swollen and dusky with medullary congestion. The
glomeruli showed fibrin thrombi within capillaries. Pigmented casts in
the tubules were noted and there were no vascular changes (Web
Fig. 2).
Pancreas showed focal acinar dilatation along with
features of early acute pancreatitis and terminal fat necrosis. There
was dullness of serosal aspect of the entire gastrointestinal tract. The
descending colon showed pin head to umbilicated lesions on the mucosa
covered with mucus. Peyer’s patches appeared prominent. Microscopically
the crypts were dilated with a focal pseudomembrane formation. Prominent
lymphoid follicles and edema were present in the submucosa.
Final Autopsy diagnosis was Acute interstitial
pneumonia (AIP), Pulmonary arterial hypertension with right ventricular
hypertrophy, Early bronchopneumonia with pulmonary thrombo-embolism,
Passive congestion and steatosis of Liver, Disseminated intravascular
coagulation of Kidneys, and Ischemic colitis.
Open Forum
Pediatric pulmonologist: Open lung biopsy is
considered to be the gold standard investigation for ILD as it shows the
actual pathology and narrows the etiological spectrum. However, many
times this would not be feasible for these children as they usually have
a severe respiratory failure or an associated infection. Bronchoalveolar
lavage could have given some clues - especially in identifying
infections, or an eosinophilic lung disease.
Pediatrician 1: Surfactant metabolism disorders
are likely with this kind of diffuse interstitial lung involvement with
the proliferation of type 2 pneumocytes. If lamellar bodies are found,
analysis of those structures would have pinpointed the diagnosis. The
lungs also showed hemorrhages, HLM, and fibroblast proliferation. This
could be well due to ARDS, DIC, and associated pulmonary hypertension.
However, if this is extensive, the possibility of primary pulmonary
hemosiderosis could be entertained.
Adult pulmonologist: There are lymphoid
aggregates and honey-comb like cysts. Moreover, there is only minimal
fibroblastic response. Why can’t this be a usual interstitial pneumonia
(UIP) or a non-HIV related lymphoid interstitial pneumonia (LIP) in this
case?
Pathology discussant: There was a florid type 2
pneumocyte proliferation in this case, which is not usually seen with
UIP. Lymphoid aggregates are focal and not uniform; hence LIP cannot be
labeled here. The presence of fibroblastic foci and widespread
inflammatory infiltrates are usually not seen in pulmonary hemorrhage
syndromes.
Discussion
Childhood interstitial lung diseases (ChILD) comprise
of a wide spectrum of disorders involving the lung parenchyma and the
etiology remains uncertain in majority of cases [2]. The incidence from
available studies ranges from 0.5 to 0.8 cases/ 100,000 children per
year [1]. Clinical presentation is often non-specific that often
contributes to a late clinical diagnosis of this condition. Common
clinical features include dyspnea, diffuse infiltrates in the chest
radiograph, and impaired gas exchange evidenced by hypoxemia. Acute
exacerbations can occur in children with pre-existing ILD due to an
infectious trigger, an episode of aspiration or the acceleration of
underlying disease process [7].
The index child had a clinical presentation
suggestive of ChILD and the pathology revealed the presence of
interstitial pneumonitis. No infectious or autoimmune etiologies could
be confirmed on the histopathology. AIP is characterized by the presence
of acute respiratory failure due to diffuse alveolar damage [8].
Differentiation of AIP from ARDS is difficult on histopathology alone.
Prodrome period for ARDS is usually less than a week, whereas in AIP,
the prodromal period is usually longer and depends on the etiology. The
onset of AIP is usually within 1-3 weeks and a rapid development of
respiratory failure may warrant mechanical ventilation in most of the
patients [9].
The etiology of AIP is not clearly known; however,
treatable infectious causes must be ruled out in a setting of AIP such
as bacterial sepsis, viral infections such as influenza,
cytomegalovirus, Mycoplasma, and Pneumocystis jirovecii
[7]. Rapidly progressive AIP could also occur in connective tissue
diseases such as Systemic lupus erythematosus, Juvenile dermato-myositis,
Anti-neutrophil cytoplasmic antibody-associated vasculitis, and Behcet’s
disease [10]. History, clinical features, and microbiological
investigations may reveal potential treatable causes of AIP such as
sepsis induced ARDS, systemic connective tissue disorder etc. Sometimes
drug-induced or transfusion-induced lung injury can also induce AIP like
picture. Patients with cryptogenic fibrosing alveolitis can also have an
accelerated presentation similar to the AIP, however, the lung
histopathology would reveal features of chronic interstitial pneumonia
along with changes of diffuse alveolar damage [11].
Our child did show some improvement following
initiation of glucocorticoids. No evidence for autoimmune causes like
SLE, Behcet’s disease or ANCA-associated vasculitides were identified.
The role of bronchoalveolar lavage (BAL) in a case of ChILD is to
identify infective causes or developmental anomalies. BAL can also
provide evidence for pulmonary alveolar proteinosis, sarcoidosis, or
hypersensitivity pneumonitis [12]. BAL could not be done in our child;
however, the tracheal aspirate did not identify any potential infective
etiology for the ILD. Role of genetic work up for the ILD in infants has
been emphasized in the recent literature. Sequencing for ABCA3
and SFTPC gene mutations have been recommended for the work-up
for infants with ILD who present after the neonatal period [12]. We
acknowledge the non-availability of BAL and genetic work-up in our
child.
In general, the management of ChILD involves the
empirical use of immunosuppressive therapy. However, in cases of
neonates and infants with severe ILD, underlying genetic defects must be
sorted out for the appropriate genetic counselling. Apart from
supportive care, neonates and infants with progressive ILD may also need
lung transplantation [13]. Our patient did respond to glucocorticoids
and developed relapse while tapering steroids and finally succumbed to
hospital acquired sepsis. The histopathology revealed features of AIP.
The treatment of AIP is mainly supportive, comprising of supplemental
oxygen therapy and invasive or non-invasive mechanical ventilation with
positive end-expiratory pressure along with empirical antimicrobials
until the bacterial sepsis is reliably ruled out. Early use of
corticosteroids has been advocated by many authors and is considered a
mainstream therapy in AIP [7]. Immunosuppressive drugs such as
intravenous methyl-prednisolone and cyclophosphamide have also been
reported to halt the rapid progression of the disease process in AIP
[14]. Other potential experimental therapies that might be useful in AIP
include surfactant therapy, inhaled nitric oxide and lung
transplantation [8].
Our case highlights one of the rare etiologies for
ChILD – AIP, and the etiology is probably idiopathic. Genetic work-up
could have thrown light on the surfactant metabolism disorders; however,
there was no evidence for pulmonary alveolar proteinosis in the lung
histopathology. The diagnosis of ChILD could be made with detailed
history, examination, and typical imaging features. Lung biopsy may not
be required in all cases to make the diagnosis, and, though genetic
tests are needed for confirmation of type of ChILD, treatment should not
be delayed while awaiting the results.
1. Clement A, de Blic J, Epaud R, Galeron L, Nathan
N, Hadchouel A, et al. Management of children with interstitial
lung diseases: The difficult issue of acute exacerbations. Eur Respir J.
2016;48:1559-63.
2. Clement A, Nathan N, Epaud R, Fauroux B, Corvol H.
Interstitial lung diseases in children. Orphanet J Rare Dis. 2010;5:22.
3. Clement A, Henrion-Caude A, Fauroux B. The
pathogenesis of interstitial lung diseases in children. Paediatr Respir
Rev. 2004;5:94-7.
4. Shachor Y, Schindler D, Siegal A, Lieberman D,
Mikulski Y, Bruderman I. Increased incidence of pulmonary tuberculosis
in chronic interstitial lung disease. Thorax. 1989;44:151-3.
5. Zar HJ. Chronic lung disease in human
immunodeficiency virus (HIV) infected children. Pediatr Pulmonol.
2008;43:1-10.
6. Kabra SK, Lodha R, Mehta P. 50 years of pediatric
pulmonology, progress and future. Indian Pediatr. 2013;50:99-103.
7. Taniguchi H, Kondoh Y. Acute and subacute
idiopathic interstitial pneumonias. Respirology. 2016;21:810-20.
8. Bouros D, Nicholson AC, Polychronopoulos V, du
Bois RM. Acute interstitial pneumonia. Eur Respir J. 2000;15:412-8.
9. Katzenstein AL, Myers JL, Mazur MT. Acute
interstitial pneumonia. A clinicopathologic, ultrastructural, and cell
kinetic study. Am J Surg Pathol. 1986;10:256-67.
10. Suda T, Kaida Y, Nakamura Y, Enomoto N, Fujisawa
T, Imokawa S, et al. Acute exacerbation of interstitial pneumonia
associated with collagen vascular diseases. Respir Med. 2009;103:846-53.
11. Akira M. Computed tomography and pathologic
findings in fulminant forms of idiopathic interstitial pneumonia. J
Thorac Imaging. 1999;14:76-84.
12. Wambach JA, Young LR. New clinical practice
guidelines on the classification, evaluation and management of childhood
interstitial lung disease in infants: what do they mean? Expert Rev
Respir Med. 2014;8:653-5.
13. Kurland G, Deterding RR, Hagood JS, Young LR,
Brody AS, Castile RG, et al. An official American Thoracic
Society Clinical Practice Guideline: Classification, Evaluation, and
Management of Childhood Interstitial Lung Disease in Infancy. Am J
Respir Crit Care Med. 2013;188:376-94.
14. Pratt DS, Schwartz MI, May JJ, Dreisin RB.
Rapidly fatal pulmonary fibrosis: The accelerated variant of
interstitial pneumonitis. Thorax. 1979;34:587-93.


