|
|
Case Reports Indian Pediatrics 2002; 39:197-202 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Nephrogenic Diabetes Insipidus with Intracranial Calcificatio |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Munni Ray Ashish Dixit Pratibha Singhi
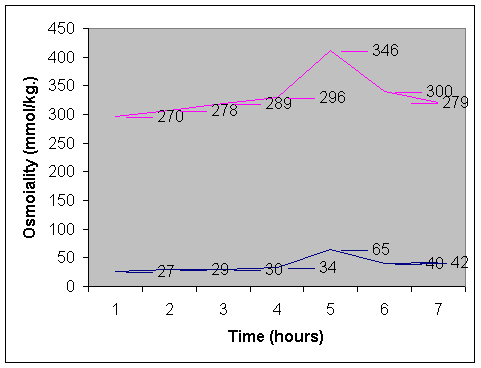
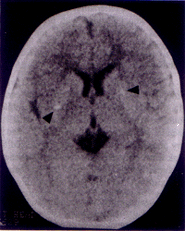
Nephrogenic diabetes insipidus (NDI) is a rare disease characterized by the failure of the kidney to respond to arginine vasopressin (AVP) because of a receptor or post receptor defect, despite raised serum concentrations of AVP. Causes of intracranial calcifications in children are numerous. Intracranial calcifica-tions with primary NDI has been recently recognized(1-3) and no such case have been reported in Indian literature. We hereby report an infant who presented with primary NDI and was detected to have intracranial calcifications. Case Report A 9-month-old infant was admitted with complaints of polydypsia, polyuria since early infancy. His mother complained that child would be irritable and would be only satisfied when he was offered water to drink. He had a very poor intake of solids, as he preferred to drink water only. His thirst was not quenched by breastfeeds alone and he consumed approximately 3-5 liters of water per day. He would pass urine frequently in large volumes. There was no history of headache, vomiting and constipation. He was born to a gravida 3 mother by full term normal vaginal delivery uneventfully. He was average sized baby at birth but had not gained weight adequately thereafter. There was no significant past history of any major illness and family history was noncontributory. He had attained his developmental milestones normally. On examination his anthropometry revealed that his length (63.5 cm) and weight (4.8 kg) were both below the third centile and his head circumference was 40 cm (<2 SD). He was euhydrated and no midline defects were identified. Fundoscopic evaluation was normal and systemic examination did not reveal any abnormality. Investigations showed serum sodium of 150 meq/L, potassium of 4.6 meq/L, blood sugar of 80 mg/dl, urea of 52 mg/dl and creatinine of 0.8 mg/dl. Complete blood count was normal. Urine specific gravity was 1005 and sugar, protein and abnormal sediments were absent. The serum osmolality was 280 mOsm/kg and urinary osmolality was 30 mOsm/kg. A water deprivation test was performed with the purpose to increase plasma osmolality to a point that is usually associated with sufficient relaease of ADH from the neurohypophysis so that the urine is maximally concentrated. Our patient had reached a maximal plasma osmolality of 297 mOsm/kg after a period of 4 hours of water deprivation when the urine was still dilute (34 mOsm/kg). Therefore he had either central or nephrogenic diabetes insipidus. Exogenous ADH was administered intravenously in the dose of 1 µg and even then the hyposthenuria was not corrected (Fig. 1). Urine culture and sensitivity was sterile. Twenty four hours urinary calcium excretion was 1.2 mg/kg/day and calcium creatinine ratio was 0.15. Renal morphology on ultrasound scanning was normal. His computed tomography of the head revealed bilateral basal ganglia calcification (Fig. 2). Intrauterine workup for the etiology of these calcifications was negative. Serum calcium was 10.5 mg/dl, inorganic phosphate was 4.5 mg/dl, alkaline phosphatase was 18 KAU and parathyroid hormone level was 50 pg/ml. Skeletal survey was normal. The Vineland Social Maturity Scale of the child revealed a social quotient of 100(4). The parents were counseled regarding adequate provision of water and a salt restricted diet for the patient. He was started on therapy with hydrochlorothiazide (3 mg/kg/d) and amiloride (0.3 mg/kg/d). On follow up of six months serum sodium was 138 meq/L and he has shown weight gain of 2.5 kg. Polyuria has reduced by as much as 40% but his urinary osmolality shows only a slight increase to 119 mOsm/L. Discussion Nephrogenic defects of urinary concentration can occur to conditions that affect primarily the action of ADH on tubular permeability to water like hypokalemia, hypocalcemia, drugs (lithium, Amphotericin B), herediatry or those which affect primarily the medullary solute concentration like acute or chronic renal failure, obstructive nephropathy or tubulointerstitial diseases(5). In the index case no secondary cause of NDI could be deciphered hence he was diagnosed as a case of primary NDI. Most cases of primary NDI have a hereditary mode of inheritence, which is usually, X linked recessive or autosomal recessive(6). As there was no family history in this case it is therefore a case of idiopathic NDI, which is clinically indistinguishable from the hereditary forms.
Fig. 2. Computed tomography of the head revealing bilateral basal ganglia calcifications.
NA - Not available. Knowledge of the pathophysiology and genetics of NDI has increased remarkably in past few years. In normal subjects AVP acts to increase the permeability of the luminal membrane in the collecting ducts to water by insertion of water channels (aquaporin 2) via V2 receptors by means of a cAMP dependent mechanism. This permits the water to flow by passive diffusion from the tubule into the hypertonic medullary interstitium in the kidney, and the urine can be concentrated. AVP induces incorporation of aquaporin 2 into the membrane via coated pits, as well as recycling via coated vesicles. Unresponsive-ness to AVP or a deficiency of AVP results in impaired incorporation and recycling of water, which means that water channels are stored in cytoplasmatic vesicles. Patients with NDI may have one of the two defects, either a V2 receptor defect (X chromosomal reces-sive) or rarely an aquaporin 2 defect(7,8). In the absence of a family history a de novo aquaporin 2 mutation is suspected(6). The incidence of pathological intracranial calcifications in 18000 pediatric computed tomograms is 1.6%(9). Calcification is most often seen in children who had prenatal in-fection with Toxoplasma gondii, Cytomegalo-virus or other inflammatory diseases. It is also found in association with or as a consequence of toxic or hypoxic damage, intracranial bleed, hypo or pseudohypoparathyroidism or metabolic and hereditary diseases like mitochondrial encephalopathy, biotinidase deficiency, etc.(10). No other underlying pathology other than NDI could be identified as a cause for the intracranial calcifications in this index case. Intracranial calcification and seizures have been noticed repeatedly in NDI particularly during infancy, when the patients are not able to compensate for fluid losses by them-selves(2). The association of two such rare conditions suggests that there is a common pathogenesis. The hypothetical sequence suggested for the development of intracranial calcification in these cases is that NDI leads to recurrent hyperosmolar dehydration. This damages the endothelial cells that exposes material with nucleating properties, e.g., mitochondria or collagen to calcium and phosphate in solution and perhaps alters calcification inhibitors such as glycosa-aminoglycans of inorganic pyrophosphate. As a consequence, calcium phosphate and other such substances are deposited particularly within or around the walls of small vessles(11). Also, in favor of this theory is the finding on brain autopsy of a patient who had NDI with intracranial calcifications where calcium deposits were found within and around blood vessels(2). It has been observed that a direct relationship exists between the severity of intracranial calcification and the duration of the condition prior to diagnosis and establish-ment of treatment(10). In our patient the calcification was mild probably because of absence of frequent attacks of dehydration. The degree of psychomotor retardation is said to be proportional to the degree of calcifica-tion. Our child had a normal developmental assessment, which corroborated with the mild calcification seen on the computed tomo-graphy. Institution of therapy early in infancy may prevent further progression of intra-cranial damage in our case, although Bagga et al. have observed that such calcification may increase despite appropriate manage-ment(12). Growth retardation as present in our index case is frequently noted finding in children with NDI. This is probably related to caloric deprivation in infants and children who have to drink water in preference to ingestion of food with an appropriate caloric content. Some believe however that it is an inherent character of disease(13). No causal treatment of this condition is available(14). Treatment focuses on the reduction of polyuria and hypernatremia to avoid dehydration and therefore preventing early and late onset complications. Low sodium diet is recommended to reduce the osmotic load that the kidney has to excrete in addition to liberal fluid intake, particularly during febrile illness. Hydrochlorothiazide alone or in combination with indomethacin was used earlier for therapy of NDI. Hypo-kalemia, renal, hemopoietic and gastro-intestinal complications were seen with these drugs. Recently long term treatment of these patients with hydrochlorothiazide and amiloride has resulted in normal growth and mental development along with better tolerance and no serious side effects have been found(15). With use of these drugs in the index case appropriate weight gain was noted and polyuria and polydypsia also improved remarkably. A review of cases published earlier presenting with nephrogenic diabetes insipidus along with intracranial calcification is cited in Table I for illustration of the association. Contributors: MR and AD carried out the clinical workup and also drafted the manuscript. PS was the consultant in charge of the patient and also supervised the drafting of the paper. She will act as the guarantor for the paper. Funding: None. Competing interests: None stated.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| References | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
![]()