Intellectual disability (ID) is the presence of
significant limitations both in intellectual functioning and adaptive
behavior as expressed in conceptual, social and practical adaptive
skills [1]. It occurs in 2-3% of the general population [2-4]. A careful
search for the cause of child’s observed delay has important
implications with reference to the risk of recurrence, prognosis,
follow-up and treatment [5]. The recent advances in fields of molecular
biology, DNA technology and prenatal diagnostic techniques have opened
up new avenues for detection and prevention of ID in families at risk.
However, there is a paucity of vital data in this area from India.
Methods
This cross-sectional study was conducted at a
tertiary care hospital in Northern India. Patients aged 3 months to 12
year who presented with ID over a period of 1 year from Feb 2010 to Feb
2011 were included. Patients were recruited consecutively from the
Genetic clinic, Outpatient department, and Pediatric wards. Our genetic
clinic is a weekly clinic. Only one or two consecutive patients who
presented first to the clinic were included as detailed evaluation
normally consumed 2-3 hours. Formal testing for developmental quotient
(DQ), intelligence quotient (IQ) and social quotient (SQ) was done by a
trained psychologist. DQ was done in children aged 3-30 months using
Developmental Assessment Scale for Indian Infants. IQ was done in
children aged 30 mo-12 yr using Binet Kulshreshtha Test. SQ was
determined in all patients using Vineland Social Maturity Scale.
Inclusion criteria used for the study were DQ/IQ <70 along with SQ <70.
Patients presenting with neuroregression and autism were also included
in the study. Previously diagnosed cases were not included. Inpatients,
who were too sick to undergo detailed workup, and those whose attendants
did not give consent were excluded. The severity of ID was graded into
mild (IQ 50-69), moderate (IQ 35-49), severe (IQ 20-34) and profound (IQ
under 20) using ICD-10 classification [6].
All children underwent detailed clinical evaluation
as per a structured proforma that included perinatal events, regression
of milestones, seizures, behavioural problems, symptoms suggestive of
inborn errors of metabolism, hypothyroidism, three generation pedigree
and social history. Examination included anthropometric, dysmorphic,
neurological and ophthalmologic assessment.
Targeted investigations were carried out on the basis
of an algorithmic approach as depicted in Fig. 1. Magnetic
resonance imaging (MRI) was done on an indicated basis in patients with
seizures/microcephaly/abnormal neurologic examination/history of
perinatal asphyxia. It was also done on a screening basis in patients
with idiopathic ID where clinical evaluation did not give any clue to
the diagnosis. Fundus examination and thyroid function tests (TFTs) were
done in patients as indicated. Dysmorphology assessment was done using
London Dysmorphology Database (LDDB) and Geneye Database. Conventional
karyotyping with GTG banding at 500nm bandwidth was done in patients
presenting with dysmorphism, multiple congenital anomalies or family
history of ID. It was not done in some patients with dysmorphism, in
whom alternative diagnosis was clinically evident (e.g.,
mucopolysaccharidoses). Molecular studies for Fragile X (FRXA and FRXE)
were done in patients with features suggestive of Fragile X syndrome and
in all males with idiopathic ID [7]. Metabolic screen (venous blood gas,
ammonia, lactate, Tandem mass spectrometry, gas chromatography) was done
in patients with history or examination suggestive of a metabolic
disorder (vomiting, seizure, abnormal odour, hypotonia/spasticity,
cataract, ataxia, hepatosplenomegaly, psychomotor regression, history of
recurrent abortions or neonatal deaths). Lysosomal enzyme assay was done
on suspicion of lysosomal storage disorder. Electroencephalogram (EEG)
was done in patients with seizures to identify the specific epileptic
syndrome. Brainstem evoked response audiometry (BERA) was done in
patients with hearing or language abnormality. If BERA could not be
done, hearing impairment was assessed by Otoacoustic Emission (OAE).
Multiplex Ligation-Dependent Probe Amplification (MLPA) was done in
patients with dysmorphism and/or multiple congenital anomalies who
demonstrated a normal karyotype. Array Comparative Genomic Hybridisation
(array CGH) was done in patients in whom MLPA was also non-contributory
but was affordable.
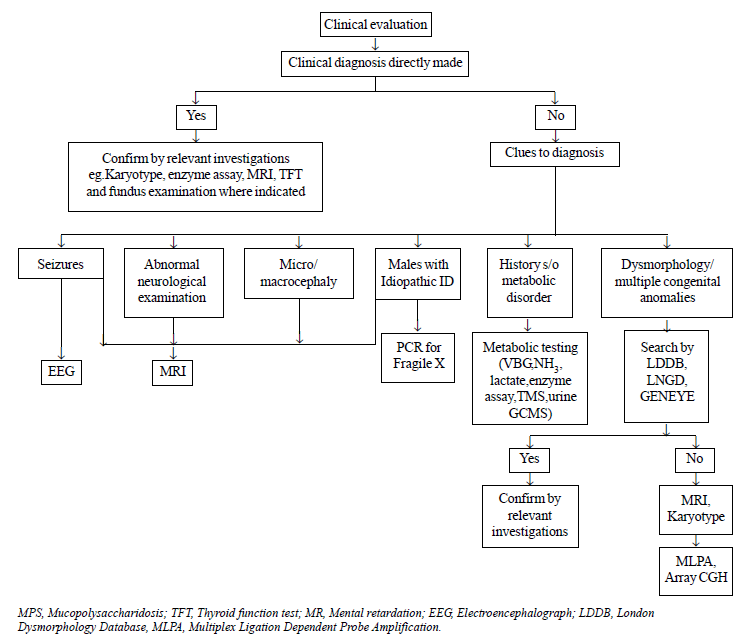 |
|
Fig. 1 Algorithm for etiological
diagnosis of intellectual disability.
|
Following the above approach, etiologically diagnosed
patients were categorized into six broad groups using the Finnish
classification [8] which is based on the timing and type of injury to
the central nervous system. Six broad groups were as follows: genetic
causes, CNS malformations, external prenatal factors, perinatally
acquired disorders (1 week before and 4 weeks after delivery),
postnatally acquired disorders and unclassified/idiopathic causes.
Genetic causes were further classified into chromosomal abnormalities,
single gene defects, contiguous gene syndromes, and recognizable
syndromes. The recognizable syndrome category included those syndromes
for which diagnosis was strongly suggested by LDDB search, but the exact
locus of genetic defect is not known. When diagnosis was ambiguous,
consultation was done with an independent trained clinical geneticist
(expert review). An etiological diagnosis was considered established
only if clinical features were supported by investigations.
Evaluation of the association between clinical
features and etiological diagnosis was made. Following clinical features
were evaluated: severity of delay, gender, neuroregression, behavioural
problems, seizures, consanguinity, family history of ID, microcephaly,
facial dysmorphism, non facial dysmorphism
(skeletal/hair/cardiovascular/gastrointestinal/urogenital anomalies) and
abnormal neurological examination (spasticity/hypotonia). Contribution
to diagnosis by history/examination/investigations was studied.
Diagnostic yield of various investigations was also determined. Written
informed consent from the primary caregivers was obtained and the study
was approved by the Institutional ethics committee.
Descriptive statistics of the study population was
generated on the basis of etiological classification used. Chi square
test or Fisher-exact test was applied to explore the bivariate
association between presence of clinical markers and determination of
etiology. For each of them, odds ratio, likelihood ratio and 95%
confidence interval were calculated and a P value of <0.05 was
taken as significant.
Results
A total of 142 consecutive children were identified
with ID over a period of 1 year. Out of the same, 13 did not give
consent, 11 did not come in follow-up, 12 inpatients were sick and 6
were found to have DQ >70 on formal evaluation. Therefore, above
patients were excluded, leaving 101 patients for final analysis. The
median age at presentation was 22 months (3months-12yr) and 65 (64.3%)
were males. Twelve (11.8%) had mild, 22 (21.7%) had moderate, 31(30.6%)
had severe and 36 (35.6%) had profound ID.
An etiological diagnosis for ID could be made in 83
patients, giving an etiological yield of 82.1%. Genetic causes
constituted the most common category accounting for 51/83 (61.4%) of the
cases followed by perinatal acquired 17 (20.4%), CNS malformations 10
(12%), external prenatal 3 (3.6%) and postnatal acquired causes 2 (2.4%)
(Table I and II). Down syndrome was the most common
cause accounting for 14.8% of all cases. Chromosomal disorders
constituted 36.5% (19/52) of genetic causes single gene 48% and
recognizable syndromes 11.5%.
TABLE I Specific Etiological Diagnosis in Genetic Disorders Category
|
Chromosomal |
19 |
|
Down syndrome |
15 |
|
Subtelomeric deletions |
1 |
|
Chromosomal aberrations by Array CGH |
3 |
|
Contiguous gene syndromes: Williams syndrome |
1 |
|
Single gene syndromes |
25 |
|
FragileX |
1 |
|
Albrights hereditary osteodystrophy |
1 |
|
Fukuyama congenital muscular dystrophy |
2 |
|
Rett syndrome |
1 |
|
Tuberous sclerosis |
1 |
|
Epileptic syndromes |
3 |
|
Congenital hypothyroid |
1 |
|
Baller gerold syndrome |
1 |
|
Metabolic |
14 |
|
Small molecule |
4 |
|
Pyruvate dehydrogenase deficiency
|
1 |
|
Malonic aciduria
|
1 |
|
Homocystinuria
|
1 |
|
Free carnitine deficiency
|
1 |
|
Large molecule |
10 |
|
Mucopolysaccharidoses
|
7 |
|
Gaucher disease
|
2 |
|
Canavan disease
|
1 |
|
Recognizable syndromes |
6 |
|
Trigonocephaly C |
1 |
|
Cardiofaciocutaneous syndrome |
1 |
|
Goldenhar syndrome |
1 |
|
Kabuki syndrome |
1 |
|
Hypomelanosis of Ito |
1 |
|
Pai syndrome |
1 |
TABLE II Etiological Profile of Other Categories
|
CNS malformations |
10 |
|
Lissencephaly
|
4 |
|
Closed lip schizencephaly
|
1 |
|
Giant open lip schizencephaly
|
1 |
|
Polymicrogyria
|
1 |
|
Corpus callosum agenesis
|
1 |
|
Dandy Walker malformation with lissencephaly
|
1 |
|
Congenital hydrocephalus
|
1 |
|
Perinatal acquired |
17 |
|
Perinatal asphyxia
|
14 |
|
Meningitis with hydrocephalus
|
2 |
|
Hypothyroid due to Mother’s TPO Ab
|
1 |
|
External prenatal |
2 |
|
Fetal valproate
|
1 |
|
Antenatal asphyxia
|
1 |
|
Postnatal acquired |
2 |
|
Brain tumor (Glioma)
|
1 |
|
Infantile tremor syndrome
|
1 |
Regression of milestones was present in 12 (11.9%) of
the total number of cases, behavioral problems in 15 (14.9%), seizures
in 39 (38.6%), consanguinity in 14 (13.9%), facial dysmorphism in 45
(44.6%), microcephaly in 30 (29.7%), family history in 16 (15.8%),
non-facial dysmorphism in 41 (40.6%) and neurological deficit in 48
(47.5%). There were 28 children with cerebral palsy. Four children had
autism. Etiological yield did not differ across gender and categories of
ID. Regression of milestones (P=0.007) and behavioral problems (P=0.025)
were significant negative predictors of etiology (Web Table I).
Visual and hearing deficit were found in 29 (28.7%)
and 23 (22.7%) patients, respectively. Fundus was abnormal in 18
patients (17.8%). Hypothyroidism was detected in 11(10.8%) including 2
patients with isolated hypothyroidism (one with congenital
hypothyroidism and another due to mother’s thyroid peroxidase
antibodies), 6 with Down syndrome, 1 with Albright hereditary
osteodystrophy, 1 with 18q subtelomeric deletion and 1 in a child with
birth asphyxia.
Diagnostic yield of investigations: Karyotype
yielded diagnosis in 15 out of 33, metabolic screen 14/34, LDDB search
8/26, Fragile X 1/4, MLPA 1/10, array CGH 3/5.MRI was done in 73
patients. It was abnormal in 52/73(71.2%), but findings consistent with
a specific etiology were found in 29/73(39.7%) patients. When done on a
screening basis, diagnostic yield was 8% (2/25) and when done on an
indicated basis yield significantly increased to 56.2% (27/48).Diagnosis
was made on the basis of investigations alone in 21/83 (25.3%) patients
and it helped in confirming the clinically suspected diagnosis in 41/83
(49.3%). Overall, it contributed to diagnosis in 62/83 (74.6%). History
gave a clue to diagnosis in 25/83 (31%), examination in 43/83 (51.8%).
Discussion
We report a study from a tertiary care-setup in
Northern India with the aim of establishing an etiological diagnosis
using relevant investigations in an algorithmic manner. An etiological
diagnosis could be assigned to 83 of 101 patients which accounts for a
high yield of 82.1%. History and examination alone contributed to
approximately 25% of the diagnosis. In another 50%, they gave important
clues for further investigation and in the remaining 25%; diagnosis was
established solely by investigations.
Although the etiologic yield has varied widely in
studies [9-22] from 20 to 86% depending on the definition used for ID,
the population studied (whether from a developmental neurology unit or a
genetic unit), the extent of diagnostic workup and the technological
advances over time, a yield as high as 80% has not yet been reported in
Indian literature except in a study by Balasubramanian, et al.
[19]. They reported a yield of 86% in a cohort of patients with ID
without autism. This may be due to an overall lack of comprehensive
Indian studies on the concerned topic and probably also due to the lack
of resources essential for establishing the diagnosis in some genetic
disorders. The most recent study by Jauhari, et al. [22] with an
etiologic yield of 54.1% in 122 patients has been reported from a
pediatric neurology setting with a different cohort and diagnostic
armamentarium. A study done recently by Tikaria, et al. [21] has
reported a diagnostic yield of 73%, but they restricted their population
to less than 5 year olds. Therefore, the results from the above studies
may not be generalizable in contrast to our study where we enrolled
children of all ages from both pediatric inpatient and outpatient
department (including the genetic clinic), with/without autism.
Moreover, our study used wider range of genetic investigations like
array CGH and MLPA, which have not been used in previous studies.
With the use of sophisticated tests, we found that
genetic causes represented the most common cause of ID. Our etiological
spectrum resembles that in most of the Western studies and also the
study by Tikaria, et al. [21] with a similar design unlike the
study by Jauhari, et al [22] where perinatal causes constituted
the most common category. This may be explained by the population
characteristics as children were enrolled from a neurology clinic.
Features like microcephaly, dysmorphism and focal
neurological deficit have been shown to be predictors of etiological
yield in most of the studies [13,16, 20,22]. This is in contrast to our
study where none of the above features were found to be significant. The
reasons for this disparity are not clear. It might be explained by the
characteristics of our undiagnosed group which was very small compared
to the diagnosed group (18 versus 83); therefore the two groups
were not comparable. Of the undiagnosed group, 44% children were
dysmorphic and did not identify with any known syndrome. They could have
represented the new genetic disorders resulting from high prevalence of
consanguinity in our patient population. However, array CGH, which might
have given a clue to diagnosis, could not be carried out in these
patients due to financial reasons.
MRI when done on a screening basis, was abnormal in
44% but yielded a specific diagnosis in only 8% of those who were
screened. This is in accordance with AAP recommendations [23] that
although MRI is often useful in the evaluation of a child with ID, it is
not a mandatory study and has a higher diagnostic yield when indications
exist. In almost all the investigations, yield was high except MLPA and
EEG. EEG was done in all patients with seizures but it helped in making
a specific diagnosis of epileptic syndromes in only 3/39(7.6%). MLPA
detects subtelomeric rearrangements, too small to be detected by
conventional cytogenetic analysis. It is based on PCR amplification of
ligated probes hybridized to chromosome ends. It was done in ten
patients with dysmorphism in whom karyotype was normal. It revealed 18q
subtelomeric deletion in 1 patient. In a recent study carried out by
Mandal, et al . [24], MLPA was found to be abnormal in only 3
(4.6%) out of 65 cases of idiopathic ID, thus confirming the lower
diagnostic yield of MLPA in unexplained ID.
Array CGH scans the genome with a high resolution,
for small chromosomal aberrations (gains/losses) or copy number variants
(CNV), which are not detected on conventional karyotyping [25]. In a
recent study by Shoukier, et al. [26], pathogenic CNVs were found
in 13.2% of 342 children with idiopathic ID. Due to financial issues,
array CGH could be done in only five patients with dysmorphism who had
inconclusive LDDB search, normal karyotype and normal MLPA. It revealed
causal diagnosis in three of them. If we had evaluated the entire
undiagnosed group, our estimates on the true yield of array would have
been more realistic. In the last few years, array CGH has emerged as a
first tier test in the context of a child with unexplained ID and
multiple congenital anomalies. It is also important for the continued
discovery of new genetic syndromes.
The strength of our study was a stepwise approach for
making a diagnosis using a wide array of advanced genetic investigations
in a representative population of children with ID. We admit that we
report a higher number of metabolic cases due to increased referrals to
the unit. Also, the sample size was small as the work was done over
limited period of one year. Diagnosis of uncommon syndromes like Baller
Gerold, Trigonocephaly C syndrome and Pai syndrome was made on the basis
of LDDB search only. They could not be confirmed by investigations as
they do not have a definite known genetic locus. Due to non-availability
of high resolution banding, karyotype could not pick up chromosomal
anomalies other than trisomy 21.
Hypothyroidism in (10.8%) patients with one patient
detected at 12 years of age with isolated hypothyroidism reinforces the
importance of diagnosing hypothyroidism early in life and also the need
of a country wide newborn screening program for diagnosing congenital
hypothyroidism.
It can be concluded that a stepwise approach should
be carried out for establishing the diagnosis of ID, using technological
advances over time. Further such studies are required using MLPA and
array CGH in India, to elucidate the real etiological spectrum of ID in
Indian children.
1. American Association on Mental Retardation. Mental
Retardation: Definition, classification, and systems of supports (10th
Ed). Washington D.C.: American Association on Mental Retardation; 2002.
2. Leonard H, Wen X. The epidemiology of mental
retardation: challenges and opportunities in the new millennium. Ment
Retard Dev Disabil Res Rev. 2002;8:117-34.
3. Roeleveld N, Ziehlius GA, Gabreels F. The
prevalence of MR. A critical review of recent literature. Dev Med Child
Neurol. 1997;39:125-32.
4. Shapiro BK, Batshaw ML. "Mental Retardation".
In: RE Behrman, RM Kliegman, HS Junson.Nelson Textbook of
Pediatrics, 18th edition. Philadelphia: Elsevier; 2008. p.191-6.
5. Shevell MI. The evaluation of the child with a
global developmental delay. Semin Pediatr Neurol. 1998;5:21-6.
6. World Health Organization. The ICD-10
Classification of Mental and Behavioural Disorders: Clinical
descriptions and diagnostic guidelines. Geneva: World Health
Organization; 1992.
7. Donnenfeld AE. Fragile X syndrome. Indian J
Pediatr. 1998;65:513-8.
8. Wilska ML, Kaski MK. Why and how to assess the
etiological diagnosis of children with intellectual disability and other
neurodevelopmental disorders: description of the Finnish approach. Eur J
Pediatr Neurol. 2001;5:7-13.
9. Majnemer A, Shevell MI. Diagnostic yield of the
neurologic assessment of the developmentally delayed child. J Pediatr.
1995;127:193-9.
10. Hunter AGW. Outcome of the routine assessment of
patients with mental retardation in a genetic clinic. Am J Med Genet.
2000;90:60-8.
11. ICMR Collaborating Centers and Central
Coordinating Unit.Multicentric study on genetic causes of mental
retardation in India. Indian J Med Res. 1991;94:161-9.
12. Battaglia A, Bianchini E, Carey JC. Diagnostic
yield of the comprehensive assessment of developmental delay/ mental
retardation in an institute of child neuropsychiatry. Am J Med
Genet.1999; 82:60-6.
13. Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz.
Etiologic yield of subspecialist’s evaluation of young children with
global developmental delay. J Pediatr. 2000;136:593-9.
14. Van Karnebeek CD, Sceper FY, Abeling NG, Alders
M, Barth PG, Hoovers JM, et al. Etiology of Mental retardation in
children referred to a tertiary care centre: A prospective study. Am J
Ment Retard. 2005;110:253-67.
15. Kabra M, Gulati S. Mental retardation. Indian J
Pediatr. 2002;70:153-8.
16. Srour M, Mazer B, Shevell MZ. Analysis of
clinical features predicting etiologic yield in the assessment of global
developmental delay. Pediatrics. 2006;118:139-45.
17. Azeem AAA, El Ela MHA, Soliman FA, El-Din AA.
Ophthalmogenetic and epidemiological studies of Egyptian children with
mental retardation. Aust J Basic Appl Sci. 2009;3:72-9.
18. Yuksel A, Kayserili H, Yesil G, Apak MY.
Evaluation of mental retardation – Part 1: Etiologic classification of
4659 patients with mental retardation or multiple congenital abnormality
and Mental retardation. J Pediatr Neuro Sci. 2007;2:45-52.
19. Balasubramanian B, Bhatt CV, Goyel NA. Genetic
studies with children with intellectual disability and autistic spectrum
of disorders. Indian J Hum Genet. 2009;15:103-7.
20. Wong VC, Chung B. Value of clinical assessment in
the diagnostic evaluation of Global Developmental Delay using a
likelihood ratio model. Brain Dev. 2011;33:548-57.
21. Tikaria A, Kabra M, Gupta N, Sapra S,
Balakrishnan P, Gulati S, et al. Aetiology of global
developmental delay in young children: Experience from a tertiary care
centre in India. Natl Med J India. 2010;23:263-8.
22. Jauhari P, Boggula R, Bhave A, Bhargava R, Singh
C, Kohli N, et al. Aetiology of intellectual disability in
paediatric outpatients in Northern India. Dev Med Child Neurol.
2011;53:167-72.
23. Moeschler JB, Shevell MI. Clinical genetic
evaluation of the child with mental retardation or developmental delays.
Pediatrics. 2006;117:2304-16.
24. Mandal K, Boggula VR, Borkar M, Agarwal S, Phadke
SR. Use of Multiplex Ligation- Dependent Probe Amplification (MLPA) in
screening of subtelomeric regions in children with idiopathic mental
retardation. Indian J Pediatr. 2009; 76:1027-31.
25. Edelmann L, Hirschhorn K. Clinical utility of
array CGH for the detection of chromosomal imbalances associated with
mental retardation and multiple congenital anomalies. Ann N Y
Acad Sci. 2009;1151:157-66.
26. Shoukier M, Klein N, Auber B, Wickert J, Schröder J, Zoll B,
et al. Array CGH in patients with developmental delay or
intellectual disability: are there phenotypic clues to pathogenic copy
number variants? Clin Genet. 2013; 83: 53-65.


