|
|
|
Indian Pediatr 2010;47: 1015-1023 |
 |
Alpha 1 Antitrypsin Deficiency in Children
with Chronic Liver Disease in North India |
|
Narendra K Arora1,2, Shivali Arora2,
Anjali Ahuja2,
Prashant Mathur2,3,
Meenu Maheshwari1,2, Manoja K Das1,2,
Vidyut Bhatia2, Madhulika Kabra4,
Rajive Kumar5, Mona Anand5,
Ashok Kumar6,
Siddarth Datta Gupta7 and Subbiah
Vivekanandan8
From the 1International Clinical Epidemiology Network (INCLEN),
2Division of Pediatric Gastroenterology, Hepatology and Nutrition, All
India Institute of Medical Sciences (AIIMS) 3Indian Council of Medical
Research,4Division of Genetics,
Department of Pediatrics, 5Oncology laboratory, Institute Rotary Cancer
Hospital,6Department of Medicine, 7Patholgy and 8Neuro-Biochemistry, AIIMS. Financial support through Department of
Biotechnology, Government of India,
Grant no. BT/PR2278/Med/13/081/2000
Correspondence to: Dr Narendra K Arora, Executive
Director, The INCLEN Trust International, F-1/5, Okhla Industrial Area,
Phase I, New Delhi 110 020, India.
Email: nkarora@inclentrust.org
Received: May 1, 2009;
Initial review: June 2, 2009;
Accepted: December 4, 2009.
Published online: 2010 March 15.
PII: S097475590900313-1
|
|
Abstract
Objective: We attempted to determine the role of
alpha-1-antitrypsin (AAT) deficient variants as an etiologic factor for
chronic liver disease in North Indian children.
Design: This study investigated 1700 children
(682 retrospectively and 1018 prospectively) (840 CLD, 410 neonatal
cholestasis and 450 without liver disease) for AAT deficiency.
Setting: Tertiary referral center, All India
Institute of Medical Sciences, New Delhi.
Patients: Of 1250 liver disease patients, 98
(7.8%) were suspected to be AAT deficient on the basis of screening
tests (low serum AAT levels and/or absent/faint alpha-1-globulin band on
serum agarose electrophoresis and/or diastase resistant PAS positive
granules on liver biopsy).
Main outcome measures: AAT deficient Z or S
allele in suspected patients.
Results: Z or S allele was not observed on
phenotyping (1700 subjects), or with PCR-RFLP, SSCP and sequencing done
in 50 of 98 suspected AAT deficient patients. A novel mutation G-to-A at
position 333 in exon V was found in two siblings having positive
immunohistochemistry for AAT on liver biopsy, both of whom had
significant liver disease with portal hypertension.
Conclusion: In conclusion, AAT deficiency as an
etiologic factor for chronic liver disease in childhood appeared to be
uncommon in North India.
Key words: Etiology, Novel mutation, Phenotyping.
|
|
Since the first description of
Alpha-1-Antitrypsin (AAT) deficiency by Laurell and Eriksson in 1963 (1),
major advances have been made in the understanding of the genetic and
clinical aspects of this disorder. Studies done among Caucasian children
suggest that AAT deficiency is among the commonest etiological factor
associated with chronic liver disease (CLD) in childhood. The allele
frequency of Z or S deficient alleles in West varies from 0.004 to
0.1%(2-4). These individuals as well as their descendants in other parts
of the world were described to be at the highest risk.
In India, AAT deficiency is being diagnosed on the
basis of low serum AAT levels, Periodic Acid Schiff (PAS) positive
diastase resistant granules on liver histology and absent or faint alpha 1
globulin band on serum electrophoresis. Based on these criteria, up to 8%
of children were suspected to have AAT deficiency associated CLD(5-8).
However, these techniques have issues of low specificity with high false
positivity(9) and no confirmed case based on either Isoelectric focusing (IEF)
or PCR based assay has ever been reported in India. In view of this
background, we attempted to determine prevalence of AAT deficiency among
pediatric chronic liver disease patients in North India using diagnostic
tests (IEF and genotyping) that are considered gold standard.
Methods
The study (retrospective: 1991-1999, prospective:
2000-2004) was conducted in pediatric liver disease patients (up to 15
years) mostly residents of North India, at the Pediatric Gastroenterology
Clinic, Department of Pediatrics, All India Institute of Medical Sciences,
New Delhi, India The caretakers of the patients who were worked up for AAT
genotype, gave consent to participate in the study.
CLD was diagnosed on the basis of clinical,
biochemical, ultrasound picture, endoscopic evidence of varices and when
feasible liver histology. Subsequent diagnostic workup was for specific
etiologies. Neonatal cholestasis was defined as conjugated bilirubin 2 mg/dL
or more than 20% of total bilirubin (whichever is less)(10). For neonatal
cholestasis, workup was done as previously described(11). The
investigation protocol is shown in Fig.1
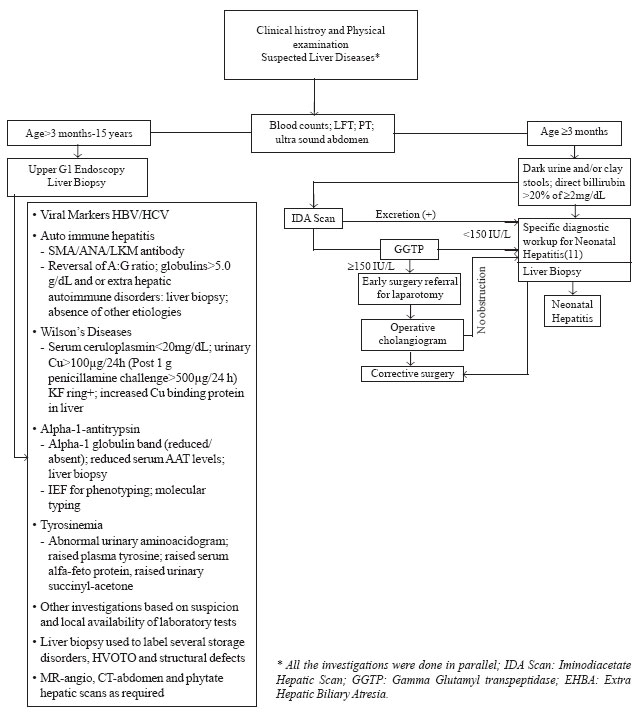 |
|
Fig.1. Investigation protocol for chronic liver disease and
neonatal cholestasis.
|
Alpha-1-antitrypsin workup
Screening: Reduction of serum levels(12) and/or a
faint or absent alpha-1-globulin band on routine gel electrophoresis
and/or histopathological evidence of PAS positive diastase resistant
granules on liver biopsy were indicators of suspected AAT deficiency
state.
Phenotyping: Isoelectric focusing (IEF) (Phast
System ®, Amersham Biosciences,
Sweden) was standardized using polyacrylamide gel slabs at a narrow pH
gradient of 4.2-4.9(13) followed by silver staining for detecting the
protein bands. Reference sera for 21 allelic variants were provided by
Prof. Magne K.Fagerhol, Oslo, Norway.
Genotypic characterization: The Pi Z (14)
and S(15) genotypic characterization was done by polymerase chain reaction
(PCR)-restriction fragment length polymorphism (RFLP) and single strand
conformation polymorphism (SSCP)(16) on DNA extracted from whole blood.
Sequencing was done commercially for confirmation.
Serum and genomic DNA from five control blood samples
were obtained from adults volunteers aged 26-49 years with no history or
evidence of CLD. Their bilirubin levels, liver enzymes, serum AAT levels
and total proteins were within normal limits. All had a PiMM phenotype.
Family screening: Father (n-43), mother
(n-48) and siblings (n-32) of the 50 suspected AAT
deficiency patients (undergoing genotypic characterization) were also
investigated for their AAT status (Pi phenotyping and mutation screening).
Statistical Analysis
The clinical and laboratory records of all patients
were entered in a database on a regular basis and appropriate consistency
checks with reference ranges put in for quality assurance purposes.
Analysis was done using "STATA" version 8.0 (STATA
Corporation, Texas, USA) statistical packages.
Results
The serum samples from 1700 patients were available for
investigating AAT deficiency. Out of these, 682 patients were recruited
retrospectively (1991-1999) and 1018 prospectively (2000-2004). A total of
840 subjects had evidence of underlying CLD and 410 had NC. Remaining 450
subjects, who had either no CLD (n-404) or NC (n-24) or were
Hepatitis B surface antigen (HBsAg) carrier (n-22) with no
histological or biochemical evidence of ongoing hepatic inflammation, were
also part of this study and labeled as "without liver disease", and served
as controls.
Etiology of CLD and NC: Among 840 patients with
definite CLD (3 months – 15 years), chronic viral hepatitis (HBV/ HCV/
mixed infection) (n-238; 28.3%) and metabolic liver diseases (MLD)
(n-159; 18.9%) were the most common etiologies. In 237 patients
(28.2%), no etiologic label could be assigned (Table I).
Table I
Etiologic Factors Associated with CLD in North Indian Children [1991-2004]
|
Etiologic Factors |
n (%) |
|
Chronic viral hepatitis |
238 (28.3) |
|
Hepatitis (HBV) |
206 (24.5) |
|
Hepatitis C (HCV) |
29 (3.4) |
|
Mixed infection (HBV/HCV) |
3 (0.3) |
|
Autoimmune hepatitis |
52 (6.2) |
|
Metabolic liver diseases |
159 (18.9) |
|
Wilson’s disease |
76 (9) |
| Glycogen
storage disease |
23 (2.7) |
|
Hereditary fructose intolerance |
14 (1.6) |
|
Lipid storage disorder |
13 (1.5) |
|
Gaucher’s disease |
6 (0.7) |
|
Bile acid metabolic defect |
6 (0.7) |
|
Tyrosinemia |
2 (0.2) |
|
Hematochromasis |
4 (0.4) |
|
Organic academia |
4 (0.4) |
|
Galactosemia |
2 (0.2) |
|
Niemann Pick disease |
2 (0.2) |
|
Byler’s disease |
2 (0.2) |
|
Indian childhood cirrhosis |
2 (0.2) |
|
Suspected MLD |
1 (0.1) |
|
AAT Deficiency |
2
(0.2) (Novel mutation) |
|
Hepatic venous outflow tract obstruction |
55 (6.5) |
|
Biliary Tract with CLD‡ |
36 (4.2) |
|
Miscellaneous |
63 (7.5) |
|
Primary Hepatic malignancy |
19 (2.2) |
|
Congenital hepatic fibrosis |
11 (1.3) |
|
Drug induced* |
8 (0.9) |
|
Celiac disease/ cystic fibrosis |
5 (0.5) |
|
Non Alcoholic Steato-Hepatitis |
4 (0.4) |
|
Other Hepatic Disorders** |
16 (1.9) |
|
Unknown etiology |
237
(28.2) |
|
Total chronic liver disease |
840 (100) |
|
No evidence of CLD† |
404 |
|
HBsAg Carriers# |
22 |
|
Incomplete workup |
136 |
|
Total Registered |
1402 |
Figures in parenthesis are percentage: ‡Gallstones (18), cholelithiasis (12), biliary cirrhosis (6)
* Histologic evidence with duration of more than 6 months; anti TB therapy(4), Valproic acid(3),
chemotherapy for acute leukemia (1); ** Histiocytosis (3); Non cirrhotic portal fibrosis (6);
Rubella (3); Kala-azar associated liver disease (4);
†Details provided in text; #No histological or biochemical evidence of liver disease.
|
A total of 410 patients fulfilled the diagnostic
criteria of NC (Table II). Obstructive causes contributed to
over one-third (141; 34.4%) cases followed by infective etiology (66;
16%). Almost one-third (30.7%) children presenting with NC could not be
assigned any specific etiology.
Table II
Etiological Factors Associated with Neonatal Cholestasis [1991-2004] [N=410]
|
Etiological Factor |
N (%) |
|
Obstructive Causes |
141 (34.4) |
|
Biliary atresia |
114 (27.8) |
|
Biliary atresia with CMV infection |
8 (1.9) |
|
Choledochal cyst |
19 (4.6) |
|
Non Obstructive Causes |
264 (64.4) |
|
Infections |
66 (16.3) |
|
CMV |
53 (13.1) |
|
Toxoplasma |
1 (0.24) |
|
Rubella |
2 (0.5) |
|
HSV |
1 (0.24) |
|
CMV/ Toxoplasma |
1 (0.24) |
|
CMV/ HSV |
1 (0.24) |
|
HBV |
5 (12) |
|
HCV |
1 (0.24) |
|
Metabolic* |
11 (2.7) |
|
Hypothyroidism (conjugated) |
22 (5.4) |
|
Bile acid metabolic defects/ Byler’s disease |
16 (3.9) |
|
Miscellaneous |
23 (5.6) |
|
Sepsis |
7 (1.7) |
|
Down syndrome |
3 (0.74) |
|
Postintestinal surgery‡ |
2 (0.5) |
|
Caroli’s disease |
2 (0.5) |
|
Alagille’s syndrome |
2 (0.5) |
|
Polycystic liver/ kidney diseases |
2 (0.5) |
|
Immunodeficiency |
1 (0.24) |
|
Hemangioendothelioma liver |
1 (0.24) |
|
Autoimmune hepatitis |
1 (0.24) |
|
Unknown Etiology |
126 (30.7) |
|
Undifferentiated |
5 (1.2) |
Figures in parenthesis are percentage: *Galactosemia(5), Hereditary Fructose intolerance (1),
Fatty Acid Oxidation Defect (1), Tyrosinemia (1), Cystic Fibrosis (1), Suspected MLD (2);
** Neonatal Cholestasis but could not be differentiated into obstructive/ non obstructive
etiologies** Excluded from the analysis; ‡Intestinal obstruction and required
corrective surgery, small bowel resection,
†Total neonates registered:529; No cholestasis 5, Incomplete workup 95 and familial
hyperbilirubinemia 19.
|
Suspected Alpha 1 antitrypsin deficiency: Ninety
eight (CLD-78; NC-20) of 1250 (7.8%) liver disease patients were suspected
to be AAT deficient on the basis of screening tests. Forty-nine liver
biopsies were possible in these 98 cases (CLD: 34; NC: 15). There were 9
biopsies with histopathological suggestion of AAT deficiency with presence
of PAS positive diastase resistant granules in the liver biopsy. Four of
these biopsies had evidence of lipofuscin material and in one PAS positive
granules were present in the cytoplasm. On follow up liver biopsies, PAS
positive granules were not observed in three of five [CLD: 1; NC: 2] study
patients. A faint/ absent alpha-1-globulin band was documented in 88
children (CLD: 72; NC: 16) and 20 children had low serum AAT levels. Five
of these 20 children with low AAT serum levels had severe malnutrition at
the time of presentation.
Underlying conditions in the group of children with
suspected AAT deficiency: CLD cases suspected to be AAT
deficient (n-78) were also worked up in parallel for other
etiologies. They had chronic viral hepatitis B/C (n-18); autoimmune
hepatitis (n-5); Wilson’s disease (n-3); other MLD’s [Gaucher’s
disease (n-1), hemochromatosis (n-1), presumed bile acid
metabolic defect (n-1). Byler’s disease (presumptive; n-1)
and hereditary fructose intolerance (n-1)]; biliary cirrhosis
(histological diagnosis, n-2); hepatic venous outflow tract
obstruction (HVOTO; n-1); miscellaneous hepatic disorders (n-5)
and unknown etiology (n-39). Similarly, NC patients (n-20)
were diagnosed as: extra hepatic biliary atresia (EHBA) (n-3);
cytomegalovirus (CMV) with neonatal hepatitis (n-3); hepatitis B
associated (n-1); suspected MLD’s (n-1); sepsis (n-1),
Down syndrome (n-1); and unknown etiology (n-10). This
etiological spectrum was very similar to the overall etiological profile
of CLD and NC patients. Clinical and biochemical profile of the patients
suspected to be AAT deficient in the initial screen (78 CLD and 20 NC) and
the rest of the liver disease patients (762 CLD and 390 NC) were similar
and did not help to differentiate the two groups.
Phenotypic characterization for AAT deficiency: IEF
was done in all 1700 children (CLD-840; NC-410; and children with no liver
disease-450). Out of 1700, 1697 had PiMM phenotype and other variants of
AAT were observed in 3 children. M1E phenotype was present in a single
patient who had unknown CLD. His mother and 3 siblings also had M1E
phenotype. Child 2 with MP phenotype had autoimmune hepatitis (ASMA and
ANA positive). Third child with MC phenotype had a history of sepsis and
had acute Hepatitis E virus (HEV) infection without CLD. The family
screening could not be done for the 2 nd
and 3rd child.
Genotypic characterization for AAT Deficiency:
Genetic workup was done for 50/98 children suspected to be AAT deficient
on screening. Two of the 98 patients were siblings and hence both were
included for complete genetic workup. Of the remaining 96 suspected AAT
deficient patients, 48 unrelated children (CLD-32, NC-16) were selected
through a computer generated random process. S or Z mutation was not found
in any of the 50 patients by PCR. Two patients (siblings) showed a shift
in band pattern in exon V on SSCP and sequencing confirmed a single base
substitution (G to A) at position 333. Index ‘1’ (younger; female) had
homozygous mutation and her sibling ‘2’ (elder; male) had hetero-zygous
base substitution. The mutation converts valine to methionine at position
333 in exon V.
Clinical details of two patients with novel mutation at
position at 333 in exon V: The index cases (1 and 2) had low serum AAT
levels (Index 1:126mg/dL and Index 2: 108mg/dL) and absence of alpha-1
globulin band on serum agarose electrophoresis. Both of them had portal
hypertension along with histological evidence of fibrosis and inflammation
without PAS positive diastase resistant granules. However,
immunohistochemistry revealed numerous rounded deposits of AAT in index 1
with homozygous mutation (Fig. 2). In index 2 (heterozygous
mutation), there were bands of fibrosis extending from central and portal
regions and immunohistochemistry was weakly positive for AAT. Neither of
them had other known etiological factor associated with liver disease.
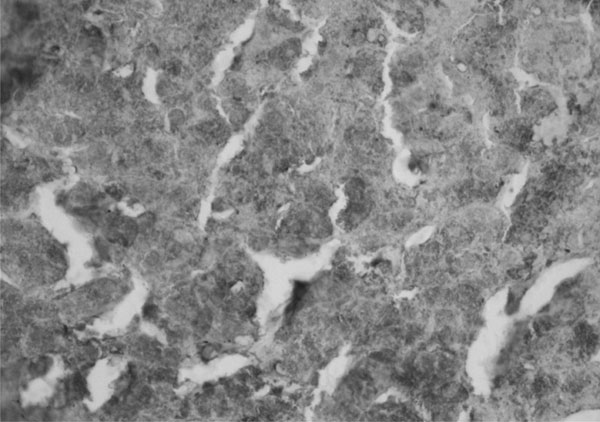 |
|
Fig. 2 Index 1(female, 120 months,
homozygous mutation at position 333 in exon V); Immunohistochemistry
(X20) showing numerous rounded alpha 1 antitrypsin (AAT) bodies. |
The siblings were born out of non-consanguineous
marriage. The mother of these patients had normal band pattern on SSCP for
both exon III and V. The father had expired at the age of 40 years in 1999
at our hospital. He was admitted with portal hypertension, grade II
hepatic encephalopathy and hepatorenal syndrome. The liver biopsy could
not be carried out but in the background of history of regular intake of
alcohol for 16 years, he was labeled as having alcoholic liver disease.
Genetic studies were not done for the father.
Quality assurance: At the inception of this study,
no expertise was available to interpret IEF gels. Thus, 10 IEF gels
including the gels containing M1E, MC and MP phenotypes and gel strip of
patient with homozygous mutation at position 333 were sent to
Alpha-1-Foundation Research Professor, University of Florida, School of
Medicine, Florida. Also, five randomly chosen DNA samples (of the patients
suspected to be alpha-1-antitrypsin deficient and chosen for detailed
genetic analysis) were sent to Genetics and IVF Institute, Virginia, USA.
Discussion
This study is an attempt at describing AAT deficiency
associated liver disease in children from India based on IEF and
genotyping. If we had based our diagnosis on low serum AAT levels and/or
absence of alpha 1 globulin band on electrophoresis and/or liver biopsy
features, 7.8% of liver disease patients would have been labeled as AAT
deficient. However, when IEF and PCR-RFLP were done, phenotypes commonly
associated with liver disease (Z and S) were not observed in any patient.
The normal variants M1E, MC and MP, detected in 3 patients have not been
described in association with pathogenesis of liver disease
(17,18). On SSCP, 2 CLD patients who were also siblings were detected to
have a novel G to A mutation at position 333. These observations indicated
the rarity of AAT associated liver disease in North Indian children.
The screening techniques have limitations particularly
in regions of low gene frequency and when the condition is rare or
extremely uncommon (19).
Furthermore, factors influencing the validity of individual screening
tests are also operating. The AAT levels may drastically reduce in
malnutrition, respiratory distress syndrome of neonates, cystic fibrosis,
nephrotic syndrome and severe liver disease (20). Five of 20 suspected
alpha-1-antitrypsin deficient subjects with low serum levels had severe
malnutrition at the time of presentation. The electrophoretic alpha 1
lipoprotein’s migratory behavior also varies with the duration of storage
of serum and with variations of intermediary lipid metabolism(1). There
are reports showing the presence of non-glycogenic PAS –positive material
in the normal as well as the abnormal liver. Fisher, et al(21)
reported a case of a patient with PiMM phenotype whose liver biopsy
sections revealed both PAS positive globules and positive
immunofluorescence. Lipofuscin granules frequently give PAS reaction,
larger granules appear coated by a PAS-positive layer. In hemochromatosis,
both in the primary idiopathic and in the secondary form associated with
anemia, the PAS reaction is strong in both Kupffer and liver cells(22). In
our study, four of nine patients with appearance like PAS positive
granules on liver biopsy had evidence of lipofuscin material. In one of
the biopsy samples, PAS positive granules were present in the cytoplasm.
On 5 follow up liver biopsies, PAS positive granules were not observed in
three [CLD: 1; NC: 2] study patients. Possibly, biliary concrements or
plugs within dilated bile canaliculi or extracellular bile deposits on the
first biopsy gave a PAS reaction, which disappeared as acute condition
settled down. These three patients did not have any residual liver disease
on follow-up. The PAS positive granules were also not observed in two
siblings with novel G to A mutation at position 333. However, AAT
inclusions were observed in both on immunohistochemistry. Thus,
specificity of all the screening tests is particularly low in regious with
rare possibility of the conditions, resulting in high possibility of false
positive tests, as was observed in our study.
In comparison to studies done among South Asian
children, the pediatric liver disease data from Caucasian population shows
several AAT alleles (homozygous and heterozygous states) which are
associated with liver diseases. In California, ZZ phenotype was found in
4.5% children with neonatal cholestasis(2) In Serbia(3), Pi*Z and Pi*S
phenotypes were found to be 15 and 3 times higher respectively in newborns
with liver disease as compared to their healthy counterparts in the
population. Absence of AAT deficient alleles in highly selected North
Indian child population with liver disease in the present study further
confirmed the findings of epidemiologic surveys done in this region(4) .
The results of the present study are also consistent to a previous study
done in India(23).
Novel mutation: Most at–risk mutations in the AAT
gene are single-base substitutions causing single amino acid modifications
in the mature protein. The number of such single base substitution
mutations is reported to be more often associated with emphysema than that
associated with liver disease. Apart from Z, a few mutations like M Malton,
Siiyama and ZBristolwere reported to be associated with liver disease
(24-26).
In the current study, IEF in both the index siblings
with mutation in exon V was similar to PiMM. Despite the mutation, the
isoelectric point of AAT molecule might not have changed and hence the
band movement was indistinguishable from PiMM allele. In an earlier
report, variant PiMM herleen (CCC to
TCC in Exon V) showed IEF pattern similar to PiMM despite the mutant AAT
allele(27). It is also important to note that both homozygous (Index 1)
and heterozygous (Index 2) states were associated with almost similar
degree of liver disease. Immunohistochemistry indicated deposits of AAT in
the hepatocytes to be denser in homozygous mutation (Index 1) than in
heterozygous mutation (Index 2). It is difficult to explain the
mechanism(s) involved in causing liver disease in Index 1 and her sibling
but role of 333 mutation in the etiology of liver disease in these two
siblings could not be ruled out completely.
For our patient population, we cannot be certain about
occasional presence of mutations in exons and introns other than exon III
and V that were not screened as part of current investigation. Such
mutations associated with liver disease are however not reported so far.
In conclusion, the study indicated that the AAT
deficiency alleles are uncommon in our population. In regions with very
low prevalence of abnormal AAT alleles, diagnosis of AAT deficiency based
on screening tests is not helpful to identify occasional patient with AAT
deficiency alleles. In strongly suspected patients, IEF and molecular
techniques should be used to diagnose the condition. We detected a single
base substitution mutation at position Val333 Met in exon V in two
siblings. The role of this novel mutation in etiology of liver disease
could not be completely ruled out. In view of our findings, we do not
recommend routine screening for AAT associated liver disease in our
region. Further study in adult emphysema patients may clarify the role of
AAT deficiency in Indians, although the possibility of it being a
significant etiologic factor appears unlikely.
Acknowledgment
We are indebted to Prof Mark Brantly, Head, Alpha 1
Research Foundation for helping us interpret IEF gels, Dr Anne Maddalena,
Genetics and IVF Institute, Virginia, USA for cross validating our DNA
samples for Z and S mutation and Dr Magne K Fagerhol, Oslo, Norway for
providing us with reference sera.
Contributors: NKA: Study design, protocol
preparation, results interpretation and manuscript editing; SA: Protocol
preparation, manuscript writing, IEF standardization, analysis of data and
other laboratory work; AA: Protocol preparation, PCR-SSCP, analysis of
data and other laboratory work; PM: Study design and patients recruitment;
MM: Laboratory support; MKD and VB: Patient screening and
recruitment; MK: Laboratory resource, genetic analysis and technical
guidance; RK and MA: Serum agarose electrophoresis standardization; AK:
Laboratory Support; SDG: Pathology; and SV: Special investigations for CLD
Funding: DBT, Govt. of India.
Competing Interests: None stated.
|
What is Already Known?
• Alpha 1
antitrypsin deficiency is prevalent in up to 8% of children with
liver disease in India based on AAT serum levels, electrophoresis
and/or liver biopsy.
What This Study Adds?
• Alpha 1
antitrypsin deficiency in children is uncommon in India. ‘Z’ or ‘S’
alleles could be altogether absent in our population.
|
References
1. Laurell CB, Eriksson S. The electrophoretic
alpha-1-globulin pattern of serum in alpha-1-antitrypsin deficiency. Scand
J Clin Lab Invest 1963; 15: 132-140.
2. Odievre M, Martin JP, Hadchouel M, Alagille D.
Alpha1-antitrypsin deficiency and liver disease in children: phenotypes,
manifestations, and prognosis. Pediatrics 1976; 57: 226-231.
3. Topic A, Jelic-Ivananovic Z, Spasojevic V, Spasic S,
Stankovic I. Distribution of alpha-1-antitrypsin phenotypes in Serbian
newborns and children with liver disease. Acta Paediatr 2002; 91: 726-727.
4. de Serres FJ. Worldwide racial and ethnic
distribution of a 1-antitrypsin
deficiency. Summary of an analysis of published genetic epidemiologic
surveys. Chest 2002; 122: 1818-1829.
5. Ramakrishna B, Date A, Kirubakaran C, Raghupathy P.
The pattern of liver disease in Indian children: a review of 128 biopsied
cases. Annals Trop Pediatr 1993; 13: 159-163.
6. Bhave S, Bavdekar A, Pandit A. Changing pattern of
chronic liver disease in India. Indian J Pediatr 1994; 61: 675-682.
7. Yachha SK, Sharma BC, Khanduri A. Current spectrum
of hepatobiliary disorders in Northern India. Indian Pediatr 1997;
34: 885-890.
8. Ganguly S, Ganguly SB. Chronic hepatobiliary
diseases in children: An etiological study. Trop Gastroenterol 1999; 20:
82-84.
9. Clausen PP, Lindskov J, Gad I, Kreutzfeldt M, Orholm
M, Reinicke V, et al. The diagnostic value of alpha
1-antitrypsin globules in liver cells as a morphological marker of alpha
1-antitrypsin deficiency. Liver 1984; 4: 353-359.
10. Moyer V, Freese DK, Whitington PF, Olson AD, Brewer
F, Colletti RB, et al. Guidelines for the evaluation of cholestatic
jaundice in infants: recommendations of the north American Society of
Pediatric Gastroenterology, Hepatology & Nutrition (NASPGHAN). J Pediatr
Gastroenterol Nutr 2004; 39: 115-128.
11. Arora NK, Kohli R, Gupta DK, Bal CS, Gupta AK,
Datta Gupta S. Hepatic technetium-99m-mebrofenin iminodiacetate scans and
serum gamma-glutamyl transpeptidase levels interpreted in series to
differentiate between extrahepatic biliary atresia and neonatal hepatitis.
Acta Pediatr 2001; 90: 975-981.
12. Corda L, Bertella E, Pini L, Pezzini A, Medicina D,
Boni E, et al. A serum level of 120 mg/dL was able to identify AAT
deficiency with a specificity of 73% and a sensitivity of 97%. Respir Med
2006; 100: 463-470.
13. Jeppsson J, Einarsson R. Genetic variants of
alpha-1-antitrypsin and hemoglobin typed by isoelectric focusing in
preselected narrow pH gradients and PHAST system. Clin Chem 1992; 38:
577-580.
14. Pamela JD. Rapid detection of alpha-1-antitrypsin
deficiency by analysis of a PCR-induced TaQI restriction site. Hum Genet
1991; 87: 742-744.
15. Andresen BS, Knudsen I, Jensen PKA, Rasmussen K,
Gregersen N. Two novel non radioactive polymerase chain reaction based
assays of dried blood spots, genomic DNA, or whole cells for fast,
reliable detection of Z and S mutations in the a 1
-antitrypsin gene. Clin Chem 1992; 38: 2100-2107.
16. Elena OP, Yon K, Agapios S, Hans V, Hans PF, Hui Z.
Detection of alpha-1-antitrypsin PiZ individuals by SSCP and DNA
sequencing in formalin fixed and paraffin embedded tissue: a comparison
with immunohistochemistry analysis. J Hepatol 2000; 32: 406-411.
17. Genz T, Martin, JP, Cleve, H. Classification of a 1-Antitrypsin
(Pi) Phenotypes by Isoelectric focusing. Hum Genet 1977; 38: 325-332.
18. Lee C, Maeng, J, Kocher, J, Lee, B, Yu, M. Cavities
of a1--antitrypsin
that play structural and functional roles. Prot Sci 2001; 10: 1446-1453.
19. Sackett DL, Haynes RB, Guyatt GH, Tugwell P. The
interpretation of diagnostic data. In: Clinical Epidemiology: A
Basic Science for Clinical Medicine. II ed, Philadelphia: Lippincot
Williams and Willkins 1991. p. 69-152.
20. Evans HE, Levi M, MandlL. Serum enzyme inhibitor
concentrations in the respiratory distress syndrome. Am Rev Resp Dis 1970;
101: 359-363.
21. Fisher RL, Sherlock S.
a-1-Antitrypsin
deficiency in liver disease: The extent of the problem. Gastroenterology
1976; 71: 646-651.
22. Luisetti M, Seersholm N.
a1-Antitrypsin
deficiency. 1: epidemiology of
a1-antitrypsin
deficiency. Thorax 2004; 59: 164-169.
23. Khanna R, Alam S, Sherwani R, Arora S, Arora NK,
Malik A. Alpha-1 antitrypsin deficiency among Indian children with liver
disorders. Indian J Gastroenterol 2006; 25: 191-193.
24. Graham A, Kalsheker NA, Newton CR, Bamforth FJ,
Powell SJ, Markham AF. Molecular characterization of three
alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi)
nullcardiff (Asp256-Val); PiMmalton (Phe51-deletion) and PiI (Arg39-Cys).
Hum Genet 1989; 84: 55-58.
25. Faber JP, Poller W, Weidinger S, Kirchgesser M,
Schwaab R, Bidlingmaier F, et al. Identification and DNA sequence
analysis of 15 new alpha 1-antitrypsin variants, including two PI*Q0
alleles and one deficient PI*M allele. Am J Hum Genet 1994; 55: 1113-1121.
26. Lovegrove JU, Jeremiah S, Gillett GT, Temple IK,
Povey S, Whitehouse DB. A new alpha-1-antitrypsin mutation, Thr-Met 85, (PIZbristol)
associated with novel electrophoretic properties. Ann Hum Genet 1997; 61:
385-391.
27. Costa X, Jardi R, Rodriguez F, Miravitlles M,
Cotrina M, Gonzalez C, et al. Simple method for
a1-antitrypsin
deficiency screening by use of dried blood spot specimens. Eur Resp
J 2000; 15: 1111-1115.
|
|
|
 |
|
