|
|
|
Indian Pediatr 2009;46: 1075-1084 |
 |
Hemolytic Uremic Syndrome |
|
Sushmita Banerjee
From the Department of Pediatrics,
Calcutta Medical Research Institute, Kolkata.
Correspondence to: Dr Sushmita
Banerjee, 9, Greek Church Row Extension, Kolkata
700026, India.
Email:
[email protected]
|
|
Abstract
Context: Hemolytic uremic
syndrome (HUS) is a severe acute disease,
sometimes with long-term sequelae. The diarrhoea-unrelated
forms are particularly associated with a poor
prognosis. The aim of this paper is to review
current evidence regarding etiology and
management, and explore methods by which the
outcome may be optimized.
Evidence acquisition: An
internet search of Medline, Medscape, MDConsult
and Cochrane databases for publications related to
HUS from 1998 onwards was performed. A review of
articles pertaining to etiopathogenesis and
management was undertaken.
Results: HUS is now
classified according to cause. New assays and gene
studies allow exact diagnosis of many of the
atypical forms. Post-exposure prevention of
diarrhoea associated HUS with vaccines and
toxin-binding agents, remains in the experimental
stages. Specific directed therapies aimed at
replacing deficient factors can improve the
outcome of atypical HUS.
Conclusions: Supportive
care remains the cornerstone of management of HUS.
The infection-unrelated forms should in addition
be treated rapidly with plasma therapy. Efforts
should be made to make an exact etiological
diagnosis in all patients, as long-term treatment
and prognosis is affected. Prevention of
diarrhea-associated HUS by improving sanitation
and proper attention to food hygiene is a
practical goal.
Key words: ADAMTS13,
Complement, Hemolytic uremic syndrome, Shiga
toxin, Thrombotic thrombocytopenic purpura.
|
|
Hemolytic
uremic syndrome (HUS) is a relatively rare disease
that can have devastating consequences. It
classically includes the triad of microangiopathic
hemolytic anemia (MHA), thrombocytopenia and renal
failure. The hallmark histopathological lesion is
thrombotic microangiopathy (TMA), characte-rized by
capillary endothelial damage and micro-vascular
formation of platelet/fibrin plugs. This induces
tissue ischemia, erythrocyte damage and consumptive
thrombocytopenia(1,2). Other than the gut and
kidney, different organs like the brain, liver and
pancreas may be affected. Thrombotic
thrombocytopenic purpura (TTP) is a similar disease,
which occurs more frequently in adults, and affects
the nervous system more than kidneys(3).
In children, diarrhea related
"typical" HUS (tHUS) is commonest (80-90%), occurs
sporadically or in epidemics, rarely recurs, and has
a relatively better prognosis(1). Atypical or
diarrhea-unrelated HUS (aHUS) is more severe,
difficult to treat, and can occur with diverse
conditions, like pneumo-coccal infections,
autoimmune disease, HIV, transplantation,
irradiation and certain drugs(4,5). Genetic forms of
aHUS, may be familial, relapsing or recurrent, and
are more commonly associated with progression to end
stage renal disease (ESRD), with high risks of
recurrence in transplants(6-8). An etiopathological
classification was recently pro-posed(9) and a
simplified version is presented in Table I.
Table 1
Classification of HUS / TTP According to Etiopathogenesis
|
Type of HUS / TTP |
Specific Cause |
|
|
|
Infection related |
Shiga toxin producing E. coli/Shigella infection |
|
Diarrhea or typical |
| |
Pneumococcal infection |
|
|
| |
HIV |
|
|
| |
Other viral or bacterial infections |
 |
|
|
Complement factor |
Factor H deficiency |
Genetic mutation (AD) |
No |
|
abnormality |
Factor I deficiency |
/Acquired antibody |
Diarrhea |
| |
Membrane cofactor protein deficiency |
|
or atypical |
| |
Factor B excessive activity |
|
|
| |
Complement 3 excessive activity |
|
|
|
ADAMTS13 deficiency |
Genetic mutation (AR), acquired antibody |
|
|
|
Cobalamin metabolism defect |
Genetic mutation (AR) |
|
|
|
Miscellaneous |
Connective tissue disease |
|
|
| |
Transplantation |
Radiation |
|
| |
Drugs |
Pregnancy |
|
| |
Malignancy |
Unknown |
|
AD: autosomal dominant inheritance; AR: autosomal inheritance inheritance.
Etiopathogenesis
Typical/Diarrhea associated/Shiga Toxin
associated HUS
Worldwide, the commonest cause of
pediatric HUS is diarrhea causing enterohaemorrhagic
E. coli (EHEC) infection(10). In developing
countries, HUS is also associated with Shigella
dysenteriae type 1 infection; however, its
incidence in India has fallen along with a reduction
in incidence of shigella dysentery(11). Rarely, HUS
can occur with E. coli urinary tract
infection(10).
Several serotypes of E. coli
are known to cause HUS, the commonest being the
serotype: 0157:H7(7). However, only about 10-15%
patients with E. coli 0157:H7 infection will
develop HUS(12). Sources of infection are milk and
animal products (incompletely cooked beef, pork,
poultry, lamb), and human feco-oral transmission(2).
Vegetables, salads and drinking water may be
contaminated by bacteria shed in animal wastes.
tHUS occurs due to bacterial
toxin production in the colon. EHEC release
verotoxin or verocytotoxin, which is structurally
and functionally homologous to Shiga toxin (Stx)
released by HUS producing strains of Shigella, and
the two terms are used synonymously. Two types of
Stx are known, Stx1 and Stx2, the latter being 400
times more virulent. EHEC adhere to and efface
intestinal cells and release Stx, which enters the
blood stream and is transported by neutrophils. Stx
binds to globotriaosyl ceramide (GB3) membrane
receptors presented on endothelial cells of kidney
and other target organs. At these sites, Stx
disrupts protein synthesis, causes endothelial cell
death and damage, induces inflammatory and
procoagulant cascades that promote microvascular
thrombosis(7,13,14). Despite producing similar
toxins, Shigella infections are associated with
higher incidence of fever, bacteremia, endotoxemia,
leukemoid reactions, severe hemolysis,
pseudomembranous colitis, and a higher fatality
rate, indicating their enteroinvasive-ness and
additional ability to cause direct cellular
injury(2).
Atypical/Non-Diarrhea Related HUS
Pneumococcal HUS
5% of all HUS and 38-43% non-diarrheal
HUS are reported in association with invasive
Streptococcus pneumoniae infection (commonly
pneumonia, empyema, meningitis, and more rarely,
pericarditis, peritonitis, bacteremia, mastoiditis,
otitis media)(5). Renal endothelial cells,
erythrocytes and platelets have a structure on their
surface called Thomsen-Friedenreich antigen (TAg).
This is normally obscured by neuraminic acid.
Pneumococci containing the enzyme-neuraminidase are
able to cleave this neuraminic acid from the cell
surface thus exposing the TAg to pre-formed anti-TAg
IgM (normally present in plasma from 6 months of
age). This leads to antigen-antibody binding,
activation of an immune cascade, with resultant,
glomerular endothelial cell damage, hemolytic
anemia, platelet aggregation and consumption, and a
fall in GFR(15,16). This TAg is also present on
hepatocytes, and hepatic dysfunction may
co-exist(17).
HUS due to Complement abnormalities
The majority of non-infection
related HUS in children is due to complement
dysregulation. Complement gene mutations are found
in 30-50% patients with aHUS(8), with 14-33% having
abnormalities in the Factor H (FH) gene, 10-15% in
membrane co-factor protein (MCP) gene and 2-13% in
Factor I (FI) gene (18-20). These genes code for
proteins that inhibit activity of complement C3b.
Deficiency causes unregulated amplification of the
alternative pathway and deposition of activated
complement on the surface of invading bacteria or
damaged self-tissue, such as apoptosed or inflamed
renal endothilial cells(18,21,22). A minority of
patients have gain in function mutations of factor B
or C3 that accelerate the activity of the
alternative pathway(23).
The majority of these genes are
situated in a cluster of complement regulatory genes
on chromosome 1q32. The mutations are generally
heterozygous, patients having reduced (but not
absent) activity of the factor, with autosomal
dominant inheritance and 50% penetration. Homozygous
and compound heterozygous mutations have also been
described, usually having a more fulminant
course(24). Autoantibodies to FH have been
identified in a few patients, some of whom in
addition have genetic deficiency of complement
factor H related proteins, CFHR1 and CFHR3.
ADAMTS13 deficiency and HUS/ TTP
ADAMTS-13 (a disintegrin-like and
metallopro-tease with thrombospondin type 1 repeats,
number 13) is an enzyme produced by stellate cells
in the liver. It acts as a von Willebrand factor (VWF)
cleaving protease, and degrades large multimeric
forms of VWF by cleaving peptide bonds. In the
deficiency of this enzyme, ultralarge multimeric
form of VWF (ULVWF) that are released by stimulated
endothelial cells circulate in plasma. Circulating
platelets spontaneously and preferen-tially bind to
ULVWF strings (rather than to smaller VWF).
Continuing platelet aggregation, ensuing TMA and
embolisation of ULVWF-platelet strings causes tissue
ischemia.
The ADAMTS13 gene is
located on chromosome 9q34. The autosomal recessive,
familial form of the disease usually seen in
children, is rare (2-3%), and occurs due to
homozygous or double heterozygous mutations of this
gene. Acquired forms of ADAMTS13 deficiency, often
associated with the presence of anti-ADAMTS13
antibodies, are more common in adults and older
children. The manifestations are more classically of
frank TTP (pentad of fever, neurological
manifestations, TMA, severe thrombocytopenia, and
relatively less severe renal dysfunction). There is
a high risk of recurrence, particularly when there
is persistence of low ADAMTS13 levels and
circulating autoantibodies during remission(3,4).
Miscellaneous causes of HUS / TTP
Abnormalities in intracellular
vitamin B12 metabolism, caused by mutations of the
cobalamin genes cause a severe HUS usually
presenting in infancy, associated with neurological
manifesta-tions, leukopenia and megaloblastic
anemia(25). HUS/TTP has been reported in association
with HIV, systemic lupus erythromatosus, and/or the
antiphos-pholipid syndrome, malignancies, radiation
and certain drugs (e.g. cyclosporine,
quinine, oral contraceptives etc.)(26). Post
transplant HUS/TTP can occur due to recurrent
disease, however de-novo disease is also seen in
both solid organ and stem cell transplantation. In
these conditions, endothelial damage leading to TMA
is postulated as the inciting factor. ADAMTS13
deficiency has been detected in some cases. Other
infections associated with HUS include viruses like
influenza, cytomegalovirus and infectious
mononucleosis, and bacteria like streptococcii and
salmonella(5).
Clinical Features
The commonest clinical
presentation of HUS is with acute pallor and
oliguria, following diarrhea or dysentery. It occurs
commonly in children between 1-5 years of age.
Hematuria and hypertension are common. Complications
of fluid overload may present with pulmonary edema
and/ or hypertensive encephalopathy. Despite
thrombocytopenia, blee-ding manifestations are rare.
Neurological symptoms like irritability,
encephalopathy and seizures may occur. Other
extra-renal manifestations include pancreatitis,
jaundice and necrosis of gut mucosa. Incomplete or
partial forms may exist(1,2,7).
Patients with aHUS have more
insidious and sometimes fluctuating symptoms at
onset, that may be preceded by viral or bacterial
illness, connective tissue disease or history of
drug intake. Family history may be present. The
degree of hypertension and duration of oligo-anuria
is greater than in tHUS. Extrarenal complications
like cerebrovascular events and pulmonary
hemorrhages, occurring due to multiorgan TMA, are
more common. Patients with genetic forms of HUS due
to complement or ADAMTS13 gene mutations, can
present in infancy, or later after a precipitating
"second hit". ESRD may ensue in the first episode or
progressive chronic kidney disease can develop with
subsequent relapses. Partial forms may occur, with
varying degrees of hemolysis, jaundice or
thrombocytopenia(4,18).
Investigations
Peripheral blood smears reveal
the presence of MHA by fragmented RBCs (schistocytes,
burr cells and helmet cells), caused by their
passage through damaged blood vessels. Platelet
counts drop due to increased consumption. The degree
of leukocytosis present has been related to a poor
outcome(27). Reticulocyte levels are high. Lactate
dehydrogenase levels are also high reflecting
increased breakdown and turnover of RBCs.
Unconjugated hyperbiliru-binemia is present due to
hemolysis. Serum haptoglobin levels are low due to
binding with released hemoglobin. The degree of
renal involvement varies and determines the increase
in blood urea, creatinine, potassium and phosphate.
In early stages, PT and APTT are normal or only
mildly deranged, differentiating from disseminated
intravascular coagulation (DIC). In some cases,
liver transaminases, pancreatic enzymes and glucose
levels may be affected. Urinalysis reveals
hemoglobinuria, hematuria and proteinuria(2,3). A
summary of investigations is given Fig.1.
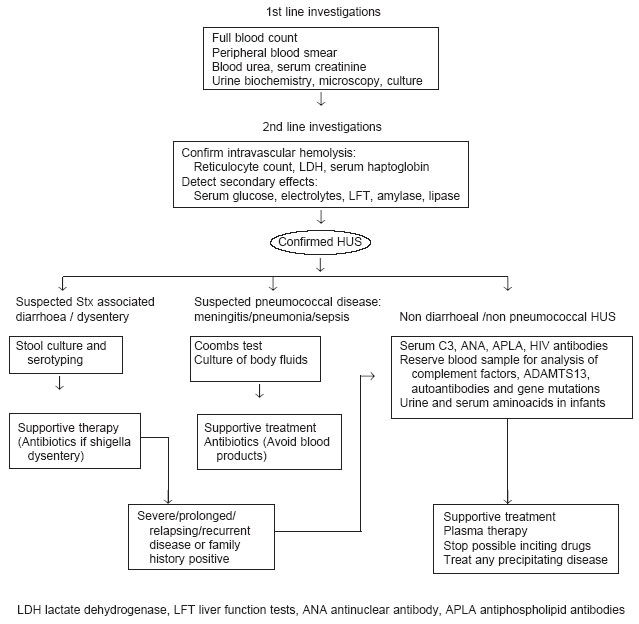 |
|
Fig.1
Management of
suspected HUS. |
Investigations to Identify Cause
In patients with dirrhea, the
identification of pathogenic EHEC or Shigella
is performed by stool culture and further serotyping
by agglutination or enzyme immunoassay(10). Shiga
toxin assays and PCR-assays have also been used for
bacteriological identification. Rarely HUS can occur
with E. coli urinary tract infection, and
urine cultures are indicated in non-diarrheal
patients. Bacteriological cultures of body fluids
(sputum/CSF/blood/pus) are indicated in suspected
pneumococcal disease.
C3 levels may be transiently low
in tHUS and persistently low in aHUS due to
complement factor deficiency. Persistently low C3
levels are commonly associated with FH or FI gene
mutations(47); however, this is not universal, and
upto 50-60% patients with demonstrable mutations
have normal C3 levels. Serum C4 levels are usually
normal(18, 19).
The direct Coombs test is
positive in over 90% of patients with pneumococcal
HUS(5); however, its specificity has not been
tested. Aminoacid chromatography of serum and urine
revealing homocystinuria, hyperhomocystinemia and
methy-malonic aciduria with low serum levels of
methionine indicates cobalamin metabolism defects.
Total serum vitamin B 12 levels are normal(25).
Autoimmune serology (ANA, anti-dsDNA, anti-phospholipid
antibodies) and HIV screening may be indicated(26).
In patients with no history of
diarrhea, blood samples for the assay of specific
complement factors (FH, FI and MCP), ADAMTS13
levels, and antibody to FH/ADAMTS13 may be taken
prior to infusion of plasma or blood products and
frozen for later analysis. Genetic studies for
mutations in complement factor genes, ADAMTS13
gene or cobalamin genes provide definitive
diagnosis. Although these studies are not available
in India, samples can be sent abroad for
analysis(28).
Renal Biopsy
Renal biopsy is not essential for
diagnosis, and often cannot be performed in the
acute stage due to thrombocytopenia. It may be
indicated in partial forms where the diagnosis is in
doubt, or in recurrent or severe disease, to confirm
the diagnosis before starting aggressive therapies.
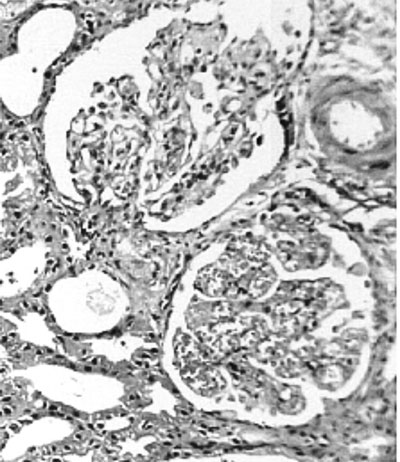
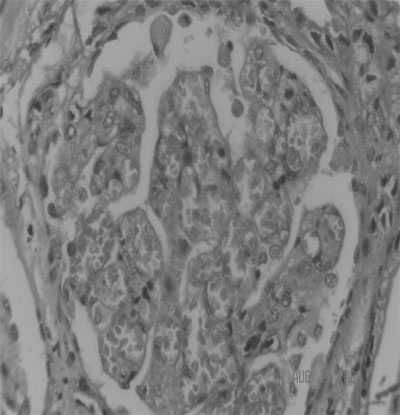
The pathognomic finding of TMA is common to all
types (Fig. 2). In tHUS, predominantly
fibrin thrombi are found in glomerular capillaries.
In contrast, in aHUS, the thrombi are made up of a
combination of fibrin, platelet and VWF clumps that
involve larger renal and interlobular arterioles,
thus causing ischemia and inflammation of larger
volumes of renal parenchyma(1). Glomerular capillary
wall thicken-ing, occlusion or narrowing of
capillary lumens, inflammation and necrosis of
endothelial cells and their detachment from the
basement membrane may be observed. Infiltration of
inflammatory cells (macrophages and neutrophils) is
seen. Tubular epithelial injury, mesangial expansion
and mesangiolysis may also occur. Cortical necrosis
is present in severe cases, and indicates a poor
outcome(2).
 |
 |
| (a) |
(b) |
|
Fig.2 Renal biopsy
of thrombotic microangiopathy in HUS.The upper
glomerulus in (a) shows ischemic contraction
and segmental mesangial proliferation. The
blood vessel at 2 O’clock position has
thickened wall with contracted lumen. RBC
casts are present in tubules. The lower
glomerulus in (a) (also shown in high power in
(b) shows endothelial swelling and capillary
loops congested with fragmented RBCs.
|
Management
Supportive Therapy
In all patients, supportive
treatment is primary. Close clinical monitoring of
fluid status, blood pressure, and neurological and
ventilatory parameters is required. Blood levels of
glucose, electrolytes, creatinine and hemogram need
frequent monitoring. The aim in early diarrhea
associated disease is to prevent dehydration and
maintain intravascular volume. The use of
antimotility therapy for diarrhea has been
associated with a higher risk of developing HUS(7).
With the onset of acute renal
failure, fluid restriction and diuretics may be
required along with salt and potassium limitation.
Antihypertensives are used to control blood
pressure. Packed cell transfusions are given to
correct significant anemia. Nutrition needs to be
maintained and parenteral nutrition may be indicated
where there is severe gut involvement. Platelet
transfusions are reserved for patients with active
bleeding, or prior to surgical procedures (like
dialysis catheter placement). Early dialyic support
is indicated if there is worsening uremia, if
electrolyte or fluid homeostasis cannot be
controlled conservatively, or if space is required
for transfusions, drugs or nutrition(1,2,7). Further
specific treatment varies according to type of HUS (Fig.1).
Antibiotics
The use of antibiotics in E.
coli associated HUS is controversial. Several
reports claimed a worse outcome with antibiotic use,
however, a meta-analysis did not support any effect
of antibiotics on the occurrence of HUS(7,29).
Antibiotics are essential for the management of
shigellosis to treat its complications and prevent
transmission.
In pneumococcal HUS, aggressive
antibiotic treatment of the primary infection is
essential. In countries where penicillin resistance
is high, vancomycin (dose adjusted for renal
involvement) should be used in addition to a 3 rd
generation cephalosporin, until sensitivity results
return.
Plasma Therapy
In aHUS due to complement factor
abnormality or ADAMTS13 deficiency, there is a
rational role for the replacement of the deficient
factor with FFP. In ADAMTS13 deficiency HUS, the
early institution of plasma therapy or
cryosupernatant, greatly improves recovery
rates(30). The use of plasma is more controversial
in HUS due to complement dysregulation. No
randomized controlled studies are available, but
recent series report a 32-72% response(8,18).
Since the specific cause of aHUS
is rarely known in the acute stage, the early use of
FFP is recommended in all non-diarrheal/non-pneumo-coccal
cases. Daily plasma infusions (10 to 20 mL/kg/day)
have been effective in some case reports, whereas in
others, plasma exchange which can deliver higher
volumes of FFP has shown better response(24,28,31).
The volume of FFP that can be infused is limited in
patients with oligo-anuria. Additionally, plasma
exchange may be more useful than infusion,
particularly in acquired forms as removal of
autoantibodies, ULVWF strings and cytokines is
facilitated.
The European Pediatric Study
Group for HUS has just published guidelines based on
‘opinion rather than evidence’, which advocates the
early use of intensive plasma exchange as primary
therapy in all patients with aHUS(28). Exchange of
1.5 times plasma volume (i.e. 60 to 75 mL/kg/day)
using FFP as replacement fluid has been recommended
in this guideline. Plasma therapy is generally
continued on a daily basis until hematological and
biochemical recovery, and then weaned gradually.
Plasma infusions at 3-weekly intervals or at the
onset of any possible precipitating illness, has
been used to prevent relapses(31).
No definite role of plasma
therapy has been documented in tHUS, although it has
occasionally been used in severe cases particularly
with neurological involvement (7,32). Plasma and
blood products may worsen the outcome in
pneumococcal HUS by providing more
anti-TAg IgM(5). In this situation, blood products
should only be used if unavoidable, RBCs should be
washed with dextran which removes 95% of plasma and
FFP should only be used if there is severe bleeding.
Miscellaneous
In infants with HUS associated
with cobalamin abnormalities, treatment with
hydroxycobalamin, oral betaine and folic acid
normalizes the metabolic abnormalities and can help
prevent further episodes(25). Removal of the
offending drug, and appropriate management of the
primary disorder is required in patients with HUS
due to drugs/HIV/connective tissue
disease/malignancy. In patients with persistent
ADAMTS13 antibodies and poor response to plasma
exchange, immunosuppressive therapy with high dose
steroids/cyclophosphamide/cyclosporin/rituximab and
splenectomy have been tried(4,26).
Outcome
The early outcome of tHUS is
relatively good in children with <5% failing to
regain renal function. In epidemics, in the acute
stage, children do better than adults. There is a
5%-10% acute mortality, mainly due to extra-renal
complications. Although the long term outcome is
better than in other forms of HUS, upto 10-30%
develop chronic kidney disease and nearly 5-10% of
these patients develop ESRD in the next 10
years(1,2). Long term follow-up is therefore,
indicated in all patients as hypertension and
proteinuria may develop after several years.
Recurrence in transplanted patients is extremely
rare.
In pneumococcal HUS, outcome
depends on degree of associated infection, and is
worst with meningitis where the mortality may be
upto 37%. The combined overall acute mortality in 73
patients, reviewed in 2007, was 12% with 75 %
requiring dialysis in the acute stage and 10%
developing ESRD on follow-up(5).
In non-infection related HUS,
upto 25% patients die in the acute phase, and 50%
progress to end stage renal failure(8). FH and FI
disease is more severe than MCP disease which may
resolve even without plasma therapy. The failure
rate of renal transplantation in FH and FI mutations
is high, with approximately 80% graft loss due to
thrombosis or recurrence(33). Combined liver and
kidney transplants have been successful in 4
patients who received intensive pre- and peri-operative
plasma exchange. In contrast, with MCP mutations,
the outcome of renal transplantation is relatively
good as MCP levels are partially replenished within
the donor kidney. The outcome of HUS due to ADAMTS13
deficiency has improved with the advent of plasma
therapy with mortality figures dropping from 80-90%
to 10-20%(4,30).
Future Developments
Ideally, the transmission of tHUS
should be prevented by improving standards of
sanitation, hygiene and food handling. Vaccines
utilizing recombinant forms of the B subunit of Stx
are under study in animals. Compounds that mimic the
structure of GB3 receptors have been synthesised (Synsorb/Starfish)
and shown to avidly bind to Stx in vitro, and in the
gut of experimental animals. Clinical trials have
not yet confirmed their efficacy in affected
children, probably because early diagnosis of Stx
production and administration of the drug would be
required to prevent the translocation of Stx to
extraintestinal sites. The use of chimeric
monoclonal antibodies to neutralize Stx and provide
passive postexposure protection are also being
studied(7).
Pneumococcal serotypes reported
to cause HUS are 1, 2F, 3, 6A, 6B, 8, 9V, 14, 19,
and 23F(15,16). The 7-valent conjugate pneumococcal
vaccine currently available in India, and included
in the universal immunization programmes of several
developed countries, contains serotypes 4, 6B, 9V,
14, 18C, 19F, and 23F. It remains to be seen whether
there will be a reduction in pneumococcal HUS
prevalence in these countries. Plasma concentrates
of Factor H and ADAMTS13, complement inhibitors and
recombinant active forms of ADAMTS13 are being
developed and may soon be available for clinical
use(31).
Acknowledgments
Renal biopsy slides were provided
by Dr SM Nadeem, Department of Pathology, Wockhardt
Kidney Institute, Kolkata.
Funding: None.
Competing interests: None
stated.
|
Key Messages
• Good sanitation and
maintenance of food hygiene can prevent
diarrhea associated HUS.
• Supportive care with
early dialysis support remains the cornerstone
of management.
• Non-infective atypical
HUS should be treated rapidly with plasma
therapy.
• Efforts should be made to make an
etiological diagnosis in cases of atypical HUS
as treatment and prognosis is affected.
|
References
1. Blackal DPl, Marques M.
Hemolytic uremic syndrome revisited-Shiga Toxin,
factor H and fibrin generation. Am J Clin Pathol
2004; 121: S81-S88.
2. Ray PE, Liu XH. Pathogenesis
of shiga toxin-induced hemolytic uremic syndrome
Pediatr Nephrol 2001;16: 823–839.
3. Sadler JE, Moake JL, Miyata T,
George JN. Recent advances in thrombotic
thrombocytopenic purpura. Am Soc Hematol Educ
Program 2004; 407-423.
4. Loirat C, Girma JP,
Desconclois C, Coppo P, Veyradier A. Thrombotic
thrombocytopenic purpura related to severe ADAMTS13
deficiency in children. Pediatr Nephrol 2009;
24:19-29.
5. Copelovitch L, Kaplan BS.
Streptococcus pneumoniae-associated hemolytic uremic
syndrome. Pediatr Nephrol 2008; 23: 1951-1956.
6. Loirat C, Niaudet P. The risk
of recurrence of hemolytic uremic syndrome after
renal transplantation in children. Pediatr Nephrol
2003; 18: 1095-1101.
7. Scheiring J, Andreoli SP,
Zimmerhackl LB. Treatment and outcome of shiga-toxin-associated
hemolytic uremic syndrome. Pediatr Nephrol 2008; 23:
1749-1760.
8. Caprioli J, Noris M, Brioschi
S, Pianetti G, Castelletti F, Bettinaglio P, et
al. Genetics of HUS: the impact of MCP, CFH and
IF mutations on clinical presentation, response to
treatment, and outcome. Blood 2006; 108:
1267-1279.
9. Besbas N, Karpman D, Landau D,
Loirat C, Proesmans W, Remuzzi G, et al. A
classification of hemolytic uremic syndrome and
thrombotic thrombocytopenic purpura and related
disorders. Kidney Int 2006; 70: 423-431.
10. Taylor CM. Enterohaemorrhagic
Escherichia coli and Shigella dysenteriae type
1-induced hemolytic uremic syndrome. Pediatr Nephrol
2008; 23: 1425-1431.
11. Ajjampur SS, Rajendran P,
Ramani S, Banerjee I, Monica B, Sankaran P, et al.
Closing the diarrhoea gap in Indian children by the
application of molecular techniques. J Med Microbiol
2008; 57: 1364-1368.
12. Karch H. The role of
virulence factors in enterohemorrhagic Escherichia
coli (EHEC) associated hemolytic-uremic syndrome.
Semin Thromb Hemost 2001; 27: 207-213.
13. Karch H, Friedrich AW, Gerber
A, Zimmerhackl LB, Schmidt MA, Bielaszewska M. New
aspects in the pathogenesis of enteropathic
hemolytic uremic syndrome. Semin Thromb Hemost 2006;
32: 105-112.
14. Karmali MA. Infection by
Shiga toxin-producing Escherichia coli: an overview.
Mol Biotechnol 2004; 26: 117-122.
15. Waters AM, Kerecuk L, Luk D,
Haq MR, Fitzpatrick MM, Gilbert RD, et al.
Hemolytic uremic syndrome associated with invasive
pneumococcal disease: the United kingdom experience.
J Pediatr 2007; 151: 140-144.
16. Vanderkooi OG, Kellner JD,
Wade AW, Jadavji T, Midgley JP, Louie T, et al.
Invasive Streptococcus pneumoniae infection causing
hemolytic uremic syndrome in children: Two recent
cases Can J Infect Dis 2003; 14: 339-343.
17. Chen JP, Chen SM, Sheu JN.
Unusual manifestation of severe conjugated
hyperbiliru-binemia in an infant with Streptococcus
pneumoniae-associated hemolytic uremic synd-rome. J
Formos Med Assoc 2007; 106 : S17-22.
18. Sellier-Leclerc AL,
Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA,
Niaudet P, Guest G, et al. French Society of
Pediatric Nephrology. Differential impact of
complement mutations on clinical characteristics in
atypical hemolytic uremic syndrome. J Am Soc Nephrol
2007; 18: 2392-2400..
19. Kavanagh D, Richards A,
Fremeaux-Bacchi V, Noris M, Goodship T, Remuzzi G,
et al. Screening for complement system
abnormalities in patients with atypical hemolytic
uremic syndrome. Clin J Am Soc Nephrol 2007; 2:
591-596.
20. Saunders RE, Goodship TH,
Zipfel PF, Perkins SJ. An interactive web database
of factor H-associated hemolytic uremic syndrome
mutations: insights into the structural consequences
of disease-associated mutations. Hum Mutat 2006; 27:
21-30.
21. Kavanagh D, Richards A,
Atkinson J. Complement regulatory genes and
hemolytic uremic syndromes. Annu Rev Med 2008; 59:
293-309.
22. Atkinson JP, Goodship TH.
Complement factor H and the hemolytic uremic
syndrome. J Exp Med 2007; 204: 1245-1248.
23. Caprioli J, Remuzzi G.
Complement hyperactivation may cause atypical
haemolytic uraemic syndrome: gain-of-function
mutations in factor B. Nephrol Dial Transplant 2007;
22: 2452-2454.
24. Cho HY, Lee BS, Moon KC, Ha
IS, Cheong HI, Choi Y. Complete factor H
deficiency-associated atypical hemolytic uremic
syndrome in a neonate. Pediatr Nephrol 2007; 22:
874-880.
25. Sharma AP, Greenberg CR,
Prasad AN, Prasad C. Hemolytic uremic syndrome (HUS)
secondary to cobalamin C (cblC) disorder. Pediatr
Nephrol 2007; 22: 2097-2103.
26. Copelovitch L, Kaplan BS. The
thrombotic microangiopathies. Pediatr Nephrol 2008;
23: 1761-1767.
27. Exeni RA, Fernández GC,
Palermo MS. Role of polymorphonuclear leukocytes in
the patho-physiology of typical hemolytic uremic
syndrome. ScientificWorld J 2007; 7: 1155-1164.
28. Ariceta G, Besbas N, Johnson
S, Karpman D, Landau D, Licht C, et al. The
European Pediatric Study Group for HUS. Guideline
for the investigation and initial therapy of
diarrhea-negative hemolytic uremic syndrome. Pediatr
Nephrol 2009; 24: 687-696.
29. Safdar N, Said A, Gangnon RE,
Maki DG. Risk of hemolytic uremic syndrome after
antibiotic treatment of Escherichia coli O157:H7
enteritis. A meta-analysis. JAMA 2002; 288:
996–1000.
30. Mannucci PM. Thrombotic
thrombocytopenic purpura and the hemolytic uremic
syndrome: much progress and many remaining issues.
Haematologica 2007; 92: 878-880.
31. Loirat C, Noris M,
Fremeaux-Bacchi V. Complement and the atypical
hemolytic uremic syndrome in children. Pediatr
Nephrol 2008; 23: 1957-1972.
32. Michael M, Elliott EJ, Ridley
GF, Hodson EM, Craig JC. Interventions for hemolytic
uraemic syndrome and thrombotic thrombocytopenic
purpura. Cochrane Database Syst Rev 2009; 21:
CD003595.
33. Loirat C, Fremeaux-Bacchi V. Hemolytic uremic
syndrome recurrence after renal transplantation.
Pediatr Transplant 2008; 12: 619-629.
|
|
|
 |
|
