|
|
Case Reports Indian Pediatrics 2007;44:933-936 |
||||
|
CINCA Syndrome |
||||
|
Chetna Khemani From the Pediatric Rheumatology Clinic, Jaslok Hospital & Research Center, Mumbai 400 016.
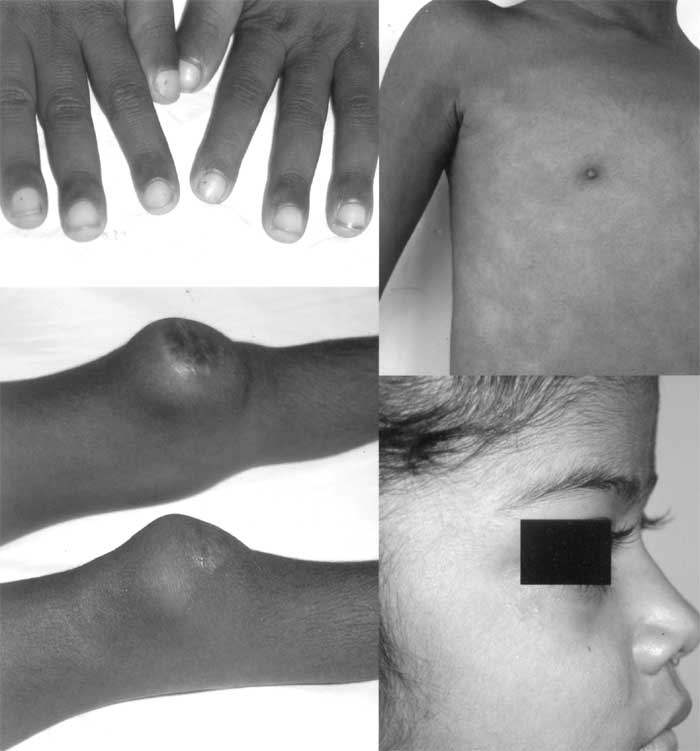
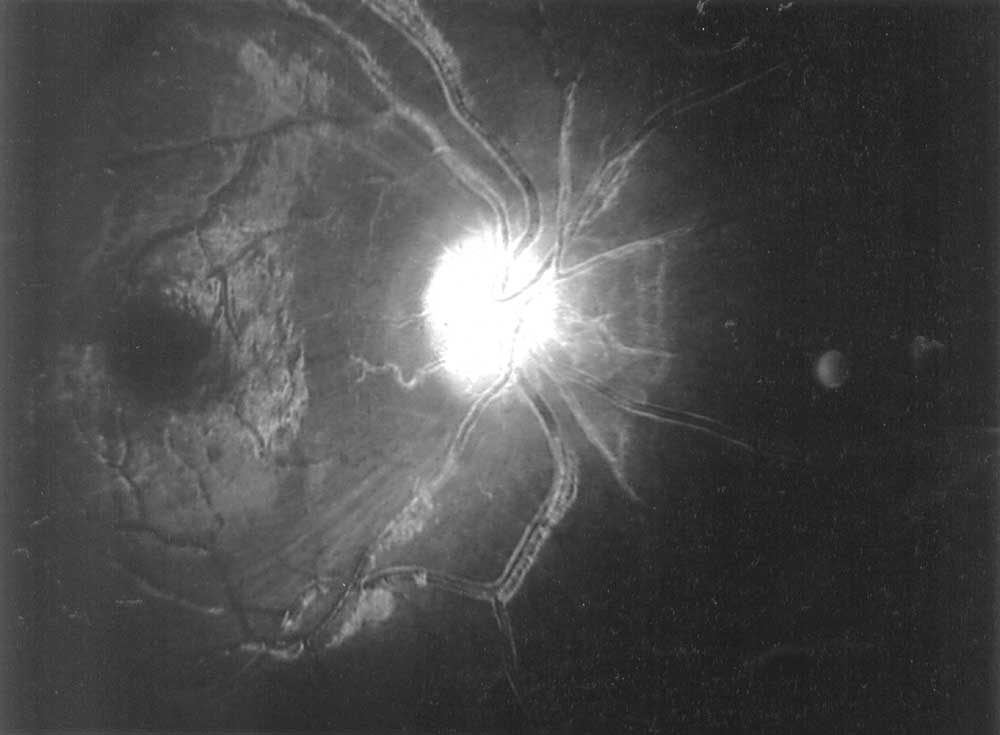
Abstract: Key words: CINCA syndrome, CIAS1 gene, retinal vasculitis. Chronic infantile neurological, cutaneous, articular (CINCA) syndrome or neonatal-onset multisystem inflammatory disease (NOMID) is a rare, genetic, autoinflammatory disorder associated with periodic fever characterized by seemingly unprovoked inflammation, in the absence of autoimmune or infective causes(1,2). It is caused by a mutation in the CIAS1 (Cold induced auto inflammatory syndrome-1) gene situated on chromosome 1(1,2). One case of an inherited inflammatory disease in a kindred of north Indian descent has been reported in world literature(3). We report the first case of CINCA syndrome in Indian literature. Case Report A 7-year-old girl was born, of a non-consanguineous marriage, by a normal delivery. A generalized urticarial rash was noticed since the second day of life. Throughout her infancy she was extremely irritable and resisted movement of her extremities. At 10 months of age, she developed recurrent febrile episodes with exacerbations of rash during these episodes. She had a delay in gross motor milestones and history of red eyes of 6 months duration. She had sparse clinical records with multiple prescriptions of analgesics, local ointments, antibiotics and intermittent oral steroids. The patient’s parents and 2 siblings were asymptomatic. Her clinical picture consisted of failure to thrive (Wt 13.5 kg, Ht 102 cm, both <3rd percentile for age), pallor, lymphadenopathy, fine conjunctival nodules, generalized nodular urticarial pruritic rash, clubbing, frontal bossing, saddleback nose, left elbow arthritis with bilateral patellar overgrowth (Fig 1). Fundoscopy showed retinal vasculitis (Fig. 2). Systemic examination was unremarkable.
Hemogram revealed Hb of 6.9 g/dL, TLC 21,900 cells/cu mm, DLC N 82%, L 15%, E 2%, M 1%, platelet count 6.78 lakhs/cumm and ESR 110 mm/h. Urine routine was normal. Skin biopsy showed atrophic epidermis and dermal inflammation with perivascular neutrophilic infiltration with no granulomas. Serum angiotensin converting enzyme levels and chest radiograph were normal. The lower limb X-rays revealed enlarged deformed femora, patellae and tibia with valgus deformity. The patellae had heterogeneous ossification. With the clinical profile suggestive of CINCA syndrome and radiological findings compatible with this diagnosis she was investigated further. CSF was normal and MRI of brain revealed cerebellar atrophy. Brainstem evoked auditory response was normal. Sequencing of C1AS1 gene amplified from genomic DNA isolated from peripheral blood leukocytes revealed F309S mutation in exon 3 on chromosome 1. She had a de novo mutation not found in her parents. She was started on prednisolone at 2 mg/kg/day with slow tapering over 12 months to 1 mg/kg/day. Azathioprine was added 3 months later at 1 mg/kg/ day progressively increased to 2.5 mg/kg/day. Since then she has had mild occasional exacerbations with bouts of fever and rash. Over a 2 year follow up her weight has improved, though she has not had optimum height gain (weight 20 kg length 105 cm). Her retinal vasculitis has not progressed and her vision remains stable. Discussion Since Prieur and Griscelli‘s first description(4) of CINCA syndrome (OMIM #607115) over 100 cases have been identified worldwide(5). Among the periodic fever syndromes are three clinically identifiable syndromes caused by mutations in the CIAS1 gene with a continuum of severity from FCAS (familial cold auto inflammatory syndrome) being the mildest, CINCA the most severe and MWS (Muckle-Wells syndrome) an intermediate phenotype(2). More than 20 mutations in the CIAS1 (also known as PYPAF1/NALP3) gene located on chromosome1 q44 have been identified(2). C1 ASI encodes for the cryopyrin protein, which is involved in inflammation, cytokine processing and apoptosis(6). The disease often starts soon after birth as in our patient and lasts the entire lifetime. Recurrent flares consisting of fever, non-pruritic urticarial rash, lymphadenopathy and hepatospenomegaly characterize the course. Articular manifestations vary from arthralgia to transient swelling without effusion to severe deforming arthropathy. Premature patellar ossification with subsequent patellar over-growth is frequent and was an important diagnostic clue in our patient. Saddle back nose, frontal bossing, short hands and feet with finger clubbing are the peculiar morphologic characteristics seen. A review of ocular manifestations in CINCA syndrome reported retinal vasculitis in 3 of the 31 patients studied(7). Neurological involvement can evolve over childhood and its delayed onset or absence as in our case has been reported in other patients(8). Differential diagnosis includes other hereditary febrile diseases, congenital infections and systemic onset of juvenile idiopathic arthritis (SOJIA). The clinical differentiating features from SOJIA would be the neonatal/ infancy onset, persistent pronounced rash, optic disc changes, progressive neurological involvement, nonspecific arthralgia and characteristic patellar bony enlargement . The other periodic fevers differ in the severity of clinical manifestations. No formal recommended treatment guidelines exist. Non-steroidal anti-inflammatory drugs and corticosteroids offer temporary relief for pain, fever and joint mobility. Various drugs including azathioprine, colchicine, cyclosporin, etanercept, infliximab, intravenous immunoglobulin, methotrexate, pencillamine, salazopyrin, thalidomide have been tried with inconsistent response. At present anakinra, an interleukin-1 (IL-1) receptor antagonist is the most promising medication for control of symptoms including neurological sequelae(9). Finances and availability were a major constraint for anakinra usage in our patient. The other deterrents for anakinra usage are the painful daily injections, potential loss of effect in the long term and exacerbation of occult infection (especially tuberculosis in our country). Azathioprine was used in our patient for its steroid sparing effect and its documented prolonged remission of retinal vasculitis(10). Acknowledgements The authors thank Drs Anne Marie Prieur and Laurence Cuisset, Groupe Hospitalier Cochin- Saint-Vincent De Paul, Paris for their advice and genetic analysis and Dr Deepak Sadarangani for his ophthalmologic inputs. Contibutors: Both the authors were involved in the conception and design, acquisition of data, analysis and interpretation of data, drafting the manuscript, critical revision of the manuscript for important intellectual content and final approval of the version to be published. Funding: Nil. Competing interests: None stated. | ||||
|
References | ||||
|
![]()