|
|
Case Reports Indian Pediatrics 2007;44:929-930 |
||
|
Fraser Syndrome: Recurrence in a Family |
||
|
Renu Singh
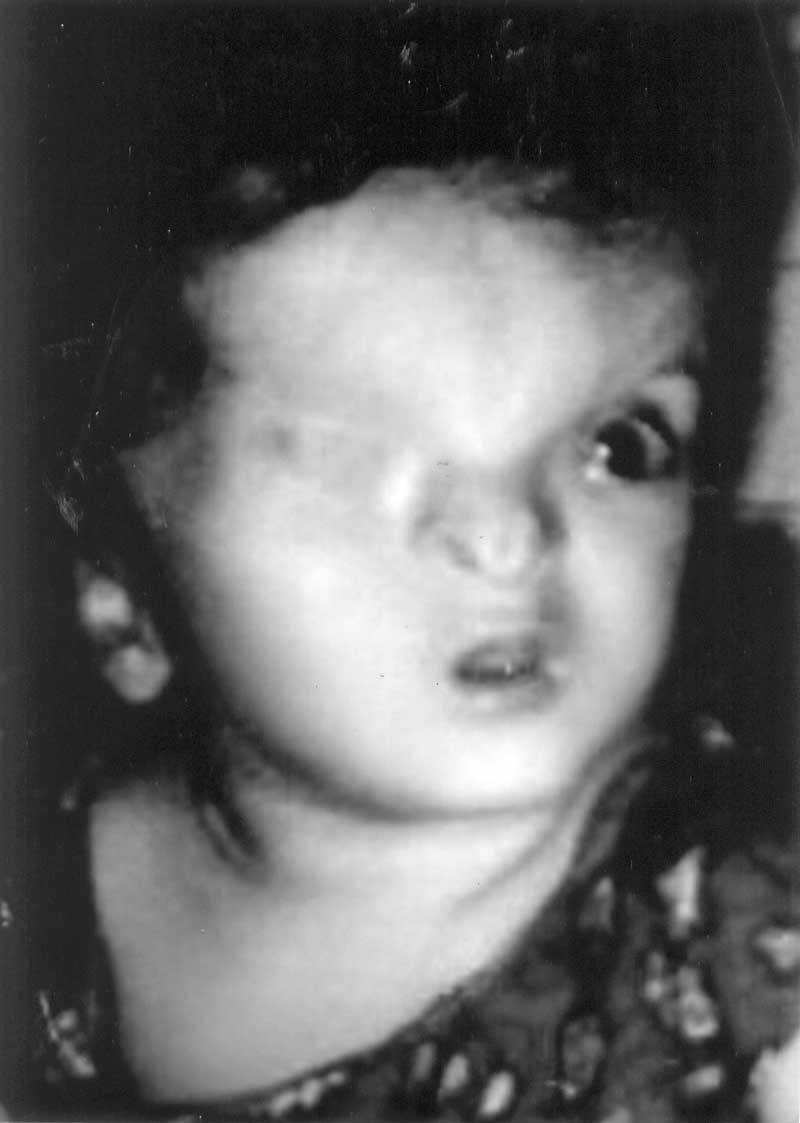
Fraser-cryptophthalmos syndrome, Recurrence. Fraser-cryptophthalmos syndrome is a rare, autosomal recessive syndrome characterized by cryptophthalmos (hidden eye), syndactyly, ambiguous genitalia, hypertelorism, a broad depressed nasal bridge, a tongue of hair extending from temple to the brow, umbilical hernia, anal stenosis and diastasis of the symphysis pubis(1,2). In this report, we describe a family with an already affected child, where the pregnant woman was referred at 24 weeks of gestation for termination of pregnancy owing to detection of severe oligohydramnios with non visualization of both kidneys on ultrasonography. The aborted fetus had findings typical of Fraser syndrome. There are no reports of molecular genetic studies for prenatal diagnosis of Fraser syndrome. Case Report A 26-year-old, second gravida, was referred at 24 weeks of gestation for termination of pregnancy owing to severe oligohydramnios with non-visualization of both fetal kidneys on ultra-sonography. The first sibling was a full term child, delivered vaginally at home. The child now one year old has multiple congenital abnormalities. The face revealed absence of eyelids and eyebrow over the right eye. It was completely covered with skin; the globe was palpable underneath (Fig.1). The nose was broad, flat with a depressed nasal bridge and a groove was present bilaterally on the nares. Ears were low set. There was microstomia with high arched palate. The external genitalia were ambiguous. The anal canal was patent. The mother’s history was otherwise unremarkable. On termination of the present pregnancy, the fetus showed multiple malformations. On examination, the face revealed absence of eyelids and eyebrows over the right eye with eye globe palpable underneath. Ears were low set with flat depressed nasal bridge, partial cutaneous soft tissue syndactyly of all fingers and toes, low set umbilicus and ambiguous genitalia.
Discussion The fetus and the elder sibling definitely fulfilled the criteria proposed by Thomas, et al.(3) to diagnose Fraser syndrome. The inheritance pattern is autosomal recessive on the basis of parental consanguinity (estimated to be as high as 15%) and multiple affected sibs born to the same parents(2).In this family two consecutive sibs were born to the same parents though the union was non-consanguineous. The karyotype of the previous affected child, phenotypically normal parents and of the fetus could not be done because of financial constraints. Fraser syndrome is rare and the FRAS1 gene has 75 exons, complicating mutation screening in affected patients. There have been no reports of molecular genetic studies for prenatal diagnosis of Fraser syndrome. Antenatal ultrasonography (level II) as early as 18 weeks of gestation has been suggested for possible diagnosis of Fraser syndrome, particularly in families with affected children(4).Fraser syndrome should be seriously considered if antenatal ultrasound shows oligo-hydramnios with contrastingly voluminous hyper-echogenic lungs(5).Prenatal diagnosis includes a combination of level II ultrasonography and fetoscopy(6), though prevention by genetic counseling would be the best approach. Contributors: RS made the diagnosis, was responsible for literature search, drafting and preparation of the manuscript. IT critically evaluated the paper and gave the final approval of the manuscript. SD was involved in the management of the patient. Funding: None Competing interests: None. | ||
|
References | ||
β-thalassemia major. Clinical and laboratory observation in 24 patients. Acta Hematol 1992; 88: 105-108. 3. Zafeiriou D, Athanasiou M, Katzos G, Economou M, Kontopoulos E. Hypoparathyroidism and intracranial calcification in b-thalassemia major. J Pediatr 2001; 138: 411. 4. Karimi M, Habibzadeh F, De Sanctis V. Hypoparathyroidism with extensive intracranial calcification in patients with b-thalassemia major. J Pediatr Endocrinol Metab 2003; 16: 883-886. 5. Kurz P, Monier-Faugere MC, Bognar B, Werner E, Roth P, Vlachojannis J, et al Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int 1994; 46: 855-861. 6. Goel A, Bhatnagar MK, V ashisha A, Verma NPS. Hypoparathyroidism with extensive intracranial calcification: a case report. Postgrad Med J 1994; 70: 913-915. |
![]()