|
|
Case Reports Indian Pediatrics 2006;43:1081-1084 |
|||||||||||||||||||||||||||||||
|
Molecular Screening of the Neutrophil Elastase Gene in Congenital Neutropenia |
|||||||||||||||||||||||||||||||
|
M. Thomas
Abstract: Congenital neutropenia is a rare hematopoietic disease, which occurs sporadically or as an auto-somal dominant inherited disorder. Pathogenesis of congenital neutropenia can now be attributed to mutations of the ELA2 gene encoding neutrophil elastase. A child with severe congenital neutropenia with a heterozygous mutation G1887A in exon 2 of ELA2 gene is reported. Key words: Molecular screening, neutropenia. Severe congenital neutropenia (SCN) or Kostmann’s syndrome is a rare disorder characterized by susceptibility to recurrent fever, severe infection and severe neutropenia (neutrophil count less than 500/mL on three occasions). SCN is caused by mutations in the gene coding for neutrophil elastase (NE), a member of the serine protease family. The primary translation product of this gene is a precursor protein of 267 amino acids synthesized at the promyelocyte stage of granulocyte differentiation(1). Molecular screening of the neutrophil elastase (ELA2) gene has identified nearly 50 different mutations most of which cluster around exons 4 and 5 corresponding to the carboxy terminus of the mature enzyme(2). Initially reported as an autosomal recessive disorder most cases reported to date have been sporadic and a few autosomal dominant cases have also been identified. We report a child with SCN who was screened for mutations in the ELA2 gene. Case report A 19 month old female child presented with a history of repeated infection since birth: neck abscess, ear lobe infection, anal fissure, otitis media with facial palsy and repeated episodes of fever. An evaluation done at four months of age revealed severe neutropenia with a hypocellular marrow and adequate megakaryocytes. Her parents were advised to do a complete blood count (CBC) weekly and during the febrile episodes (Table I) which did not show a cycling pattern. Flow cytometry studies showed normal expression of T and B lymphocyte subsets (IgG1 = 0.16, CD3 = 64.9, CD4 = 28.9, CD8 = 21.2, CD11 = 92.4, CD20 = 27.5) and cytogenetic analysis was normal. Bone marrow smear showed normo-cellular marrow with myeloid maturation arrest, focal plasmacytosis and reduced iron stores. The child was started on Inj. G-CSF (50 microgram) thrice weekly on which she showed symptomatic improvement. There was no family history of similar illness in either of her parents and no history of consanguinity. TABLE I Complete Blood Count of Patient at Diagnosis and after start of R-GCSF
ANC* absolute neutrophil countDLC* differential countWBC* White blood countHb* Hemoglobin# Patient having middle ear infection with fever at the time¶ Serial monitoring showed no cyclical depression of neutrophils
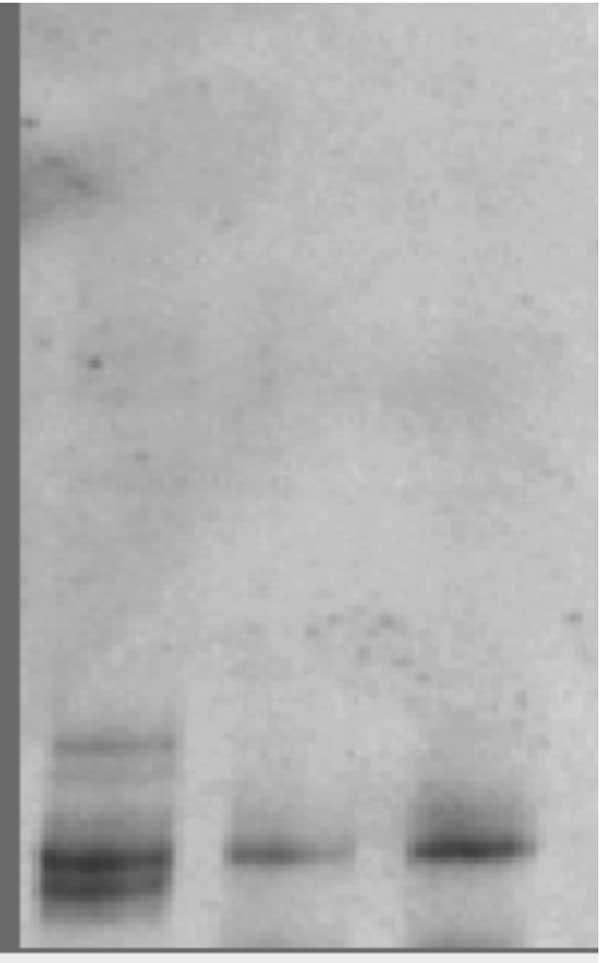
Mutation analysis After informed consent, genomic DNA from the patient and parents was extracted from peripheral blood leucocytes using phenol-chloroform method. Three fragments of DNA covering all exons of ELA2 and at least 15 bases of the flanking regions were amplified as described previously(3). PCR products were then loaded in a mildly denaturing 10% conformation sensitive gel electrophoresis (CSGE) as described previously(4) (Fig. 1). PCR products showing abnormal mobility by CSGE were sequenced using the Big Dye Terminator Cycle Sequencing kit (Applied Biosystems, Warrington, UK) on an ABI 310 genetic analyzer (PE Applied Biosystems, Foster City, CA, USA).
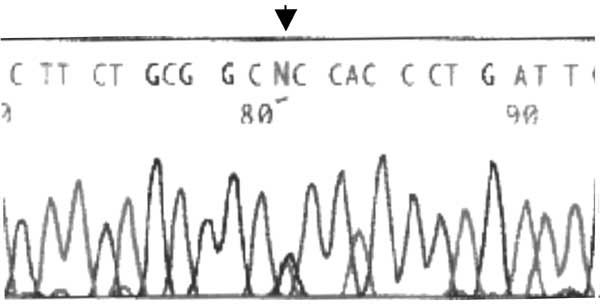
A heterozygous missense mutation G1887A in exon 2 of the gene encoding the neutrophil elastase protein was found (nucleotide nomenclature of the neutrophil elastase gene is ELA2 gene according to GenBank Y00477 sequence) (Fig 2). PCR analysis of the parents failed to detect any mutation at the identical locus. Before terming it as de novo mutation a microsatellite PCR analysis was performed for five markers (Fes, THO1, VwA, F13A1, ACTBP2)(5-7) on the patient as well as the parents sample and the ACTB P2 analysis confirmed the parentage.
Discussion Over fifty different mutations have been reported in the neutrophil elastase gene. Heterozygous mutations in the ELA2 gene coding the neutrophil elastase enzyme have been detected in most patients with SCN. In the present case, a heterozygous G 1887 A (codon 28 GCC→ACC) was detected which results in the replacement of a hydrophilic alanine residue with a hydrophobic threonine (A 28 T) at the amino terminal end of the mature protein. This has been previously reported by Dale(8) where it was associated with a very severe phenotype (severe maturational arrest). Normal genotype and phenotype in the parents suggests that this case is due to a new (de novo) mutation. However the possibility of germline mosaicism cannot be ruled out(9). However we were not able to confirm the presence of either in the present study. The biochemical mechanism by which this mutation result in neutropenia has not been elucidated but it is linked to the poor survival characteristics of the myeloid precursor cells. Aberrant processing, intracellular targeting and subsequent expression of the mutant elastase inducing premature activation of "death" pathways have been demonstrated for one of the reported mutations (G185R). Various possible substrates of the mutant enzyme include the G-CSF, G-CSF receptor, Bcl2 family members and the Notch receptors. Interaction with all the above-mentioned substrates promotes apoptosis of the differentiating granulocyte progenitors resulting in the maturational arrest(2). These patients are usually in need of supra-physiological amounts of recombinant G-CSF that probably prevents interaction of mutant elastase with these substrates. Approximately 10-15% of SCN patients harboring mutant neutrophil elastase and who require therapy with supraphysiological amounts of recombinant G-CSF have an increased predisposition to developing AML(10). Severity of neutropenia can vary in SCN and genotype-phenotype correlation needs to be established in order to determine the pathogenesis of ELA2 mutations in causing the varying phenotypes in SCN. Contributors: MT: Preparation of protocol and manuscript, genetic diagnosis of congenital neutropenia,; GJ: study and experimental design and appraisal of protocol; MC: Supervision and final approval of protocol, study design and manuscript. Funding: This study was supported by grant BT/MB/04/2004 from the Department of Biotechnology, Government of India. Competing interest: None.
| |||||||||||||||||||||||||||||||
|
References | |||||||||||||||||||||||||||||||
|
![]()