|
|
Case Report Indian Pediatrics 2003; 40:1201-1203 |
||||
|
Congenital Long QT Syndrome Presenting as Epilepsy |
||||
|
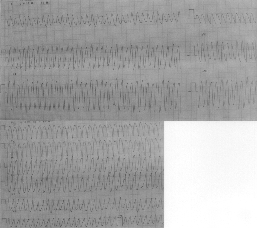
Congenital Long QT Syndrome is a genetic disorder affecting ion channel function resulting in repolarization abnormalities and an increased propensity to develop potentially lethal ventricular tachyarrhythmias. The condition may remain unrecognized for a long time in children who present with recurrent syncope, seizures or drop attacks. We report a case of congenital Long QT Syndrome masquerading as idiopathic epilepsy. Case Report A 11-year-old boy was hospitalized with a history of two seizures while at home. The first seizure had occurred while he was watching TV. He complained to his mother about a feeling of discomfort in the chest. He lost consciousness almost immediately thereafter and was noticed to develop stiffness of the extremities. The episode terminated within a few minutes spontaneously. He regained full normalcy after a short time without any significant drowsiness or neurologic deficits. However he had a second episode after half an hour that lasted longer. He was immediately brought to the hospital. There was a history of such episodes in the past, beginning at the age of 6 years. He used to have seizures about twice a year. The episodes usually occurred in the early morning while he was still asleep and would often be associated with urinary incontinence. He had been diagnosed to have idiopathic epilepsy and was being treated with carbamazepine. Three days before this admission, he had a "breakthrough seizure" and the dose of carbamazepine had been increased. He was the second child of a second degree consanguineous parentage. His elder brother is healthy. There was no history of seizures or sudden death in any close family members. He was not taking any medications other than carbamazepine. In the hospital, he had a seizure during which he was found to be pulseless. External cardiac massage was given and he was revived. An ECG taken immediately afterwards showed sinus rhythm with a prolonged QT interval. While his ECG was being monitored, he was noticed to develop a rapid monomorphic ventricular tachycardia (Fig. 1) with hypotension. Magnesium was given intravenously as a bolus followed by an infusion. Intravenous lignocaine infusion was started but there was no response and the tachycardia was terminated with a DC shock.
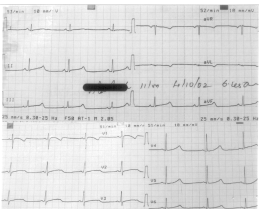
The ECG in sinus rhythm (Fig. 2) showed sinus bradycardia at a rate of 52/min with a corrected QT interval of 510 milliseconds. The T waves were broad and notched in the midprecordial leads. Serum potassium was 3.8 mmol/L, serum calcium was 4.3 mmol/L and serum magnesium was 1.6 mmol/L. Echocardiography showed no structural abnormality. Propranolol was started in a small dose. However, it led to significant bradycardia with occurrence of multiple ventricular ectopics and was hence withdrawn. Polymorphic ventricular ectopics and short runs of monomorphic ventricular tachycardia recurred over the next 24 hours despite lignocaine infusion. Considering his age, it was decided to give him a trial of oral nicorandil before implantation of a permanent pacemaker. Nicorandil was begun at a dose of 2.5 mg/d and gradually increased to 10 mg/d over two weeks. Since then he has been free of ventricular tachycardia or ventricular ectopics. Holter monitoring for 24 hours before discharge revealed no premature beats or tachyarrhythmias. Exercise testing on a treadmill also did not precipitate any arrhythmias. Screening of other family members showed normal electrocardiograms in the parents, elder brother and maternal grandmother. At last follow up eight weeks after nicorandil was started, he remained asymptomatic. The ECG showed a sinus rhythm at a rate of 85/min with a corrected QT interval of 470 msec. Discussion The Long QT syndrome may often be missed because it is not suspected in children who present with syncope, seizures or drop attacks. Cases have previously been reported in which a child treated for a long time for idiopathic epilepsy was found to have the long QT syndrome(1-4). This boy was being treated for idiopathic epilepsy for five years with about two episodes per year despite anticonvulsant therapy. However the history did not provide any clues to suspect a non-epileptic seizure. A history of onset of unconsciousness before the seizure has been suggested as a differentiating factor(5) but this could not be elicited. There was a history of premonition just preceding an attack, but this has been reported with the tachyarrhythmias associated with the Long QT syndrome also(2). The co-occurrence of seizures and a prolonged QT interval with ventricular tachycardia has been reported due to tricyclic antidepressant poisoning(6). Interestingly, familial epilepsy has been described due to abnormalities in the KCNQ family of genes that also causes long QT syndrome type I, though the specific gene is different(7,8). Though the usual tachyarrhythmia associated with the long QT syndrome has been torsades de pointes, monomorphic ventricular tachycardia can also be seen. Beta-blockers have been the mainstay of therapy, however they may worsen the situation in children with a pause dependent ventricular tachycardia by producing bradycardia. They may be combined with pacing in such situations. Identification of the underlying defects in ion channel function has led to the use of novel therapies. Nicorandil, a potassium channel activator, would theoretically benefit those with a defect in potassium channel function. Nicorandil administered either intravenously(9) or orally(10) has been shown to result in a decrease in the QT interval and abolition of repolarization alternans. The presence of a prolonged QT interval, notched T waves and sinus bradycardia along with documented ventricular tachycardia during the "seizures" confirms the diagnosis of the Long QT syndrome. This possibility should be considered by pediatricians who see a child with seizures or recurrent syncope. Contributors: JMT and CSP were responsible for the primary management of the patient. SRJS was responsible for drafting the manuscript. GS oversaw the case management and manuscript drafting. Funding: None Competing interests: None stated
| ||||
|
References | ||||
|
![]()