|
|
Editorial Indian Pediatrics 2000;37: 827-830 |
|||
|
Neonatal Cholestasis Syndrome– The Saga Continues |
|||
|
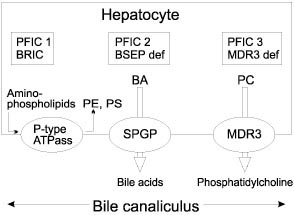
The importance of early diagnosis in a child with cholestatic jaundice has been emphasized repeatedly especially with respect to biliary atresia (BA) as shown by good results of early surgery (Kasai portoenterostomy) for this condition. Over 80% of infants with biliary atresia who underwent surgery before 60 days of age have become jaundice free compared with 20-35% for those with later surgery(1). It was clear even 20 years ago that infants who cleared their jaundice had a good prospect of long term survival with a good quality of life(2). A long-term outcome study showed a 15- year survival of 87% in infants who become jaundice free(3). The Children’s Liver Disease Foundation launched an educational program in United Kingdom in May 1993 with the help of the Department of Health to improve the outcome of hepatobiliary disease in infants. The aim of that campaign was to make certain that all babies who remain jaundiced after 2 weeks of age are tested for conjugated hyperbiliru-binemia and referred for timely specialist investigations and treatment, if positive. Such infants most commonly have disorders other than biliary atresia for which early and specific treatment is equally desirable(4). Prompt and proper investigations of these babies is important in diagnosing treatable causes, e.g., metabolic (galactosemia, tyrosine-mia), endocrinal (congenital hypothyroidism, congenital hypopituitarism), infective (malaria, congenital syphilis, toxoplasmosis, generalized sepsis, etc.), surgical (choledochal cyst, biliary atresia) and it will also be able to decrease the morbidity due to late presentation. Persistent cholestasis can lead to malabsorption leading to failure to thrive, hence these babies need more calories to maintain satisfactory growth which is very important in first few years of life. These babies have fat soluble vitamin defi-ciency states and require vitamins A, D, E, and K supplementation. They are at special risk of life threatening bleeding or brain damage from vitamin K responsive hemorrhagic disease, a risk that may be increased by the current decline in the use of parenteral vitamin K in the newborn(5). Management of such babies in a specialist referral center is the key. It has been shown that results of Kasai portoenterostomy are less satisfactory in centers operating on less than 6 cases per year(6). The French national study on prognosis of biliary atresia in the era of liver transplantation between 1986 to 1996 concluded that the overall prognosis of BA depends on the same independent prognostic factors as survival with native liver: the overall survival is better if the Kasai portoenterostomy is performed, when the child is operated early, when the anatomical pattern of BA is favorable (in order of decreasing prognosis: atresia limited to common bile duct, atresia with cyst in the liver hilum communicating with hairy intrahepatic bile ducts, atresia with gallbladder and common bile duct patent, and complete extrahepatic BA), the absence of polysplenia syndrome, and when the center has large experience in the management of BA patients(7). Since it is not possible for every medical center to upgrade its investigative facilites, there is a need for establishment of integrated services with regional and subregional set-up with exclusive diagnostic facilities, which can serve as a referral center for other units. The "Consensus Report on Neonatal Cholestasis Syndrome" has identified the following factors: delay in referral of these patients, need of early pick up and referral to a specialized center, need of early diagnosis specially for biliary atresia and other treatable causes, importance of nutrition, adoption of a uniform management protocol, and establish-ment of integrated services(8). An audit of these recommendations in few years time will be valuable. At King’s College Hospital, between 1989-1999, out of 998 cases of neonatal cholestasis syndrome, 44.4% were idiopathic neonatal hepatitis, 22% biliary atresia, 2.4% choledochal cyst, 8.1% alpha-1-antitrypsin deficiency, 4.5% Alagille syndrome, 2% progressive familial intrahepatic cholestasis, and 2.3% TPN related cholestasis. In our study there were 3 cases of CMV hepatitis (diagnosis was confirmed on CMV inclusion bodies on liver biopsy and CMV DNA positivity) whereas in the Consensus Report the incidence of CMV hepatitis was reported to be around 5.9%. We think that CMV as a cause of neonatal hepatitis is an over diagnosed clinical entity because of the ubiquitous nature of the Cytomegalovirus. The diagnosis of CMV hepatitis in our practice is entertained in the presence of CMV inclusion bodies with hepatitis on liver biopsy, high CMV DNA levels in the absence of other causes of infantile cholestasis rather than isolated CMV IgM positivity. The treatment with Ganciclovir should only be considered in immunocompromized infants or in the presence of worsening liver function tests. Once early diagnosis and management of the common conditions is achieved the need will arise to look for the less common and newly recognized conditions. Endoscopic retrograde cholangio-pancreatography (ERCP) has a special role in the diagnosis of neonatal sclerosing cholangitis, a relatively new clinical entity where liver biopsy may be compatible with BA but stools are pigmented. Another interesting technique is magnetic resonance cholangiopancreato-graphy (MRCP) but its role as a diagnostic modality to identify biliary tract pathology in pediatrics is still in infancy. In the last decade with the advancements in molecular and genetic testing, a number of genetic cholestatic syndromes have been identified. Alagille syndrome has been linked to a JAG-1 gene on chromosome 20. Numerous mutations have been reported in this gene. Progressive familial intrahepatic cholestasis (PFIC), previously known as Byler syndrome or Byler like disease has been related to mutations in hepatocellular transport system genes involved in bile formation. Three different types have been identified so far (Fig. 1).
PFIC 1 and benign recurrent intrahepatic cholestasis (BRIC) are due to mutations of the FIC 1 gene encoding a P-type ATPase protein involved in aminophospholipid transport, FIC1 gene is located on chromosome 18q 21-22. PFIC2 is due to mutations of the SPGP (sister of P-glycoprotein) gene encoding the ATP-dependent canalicular bile acid transporter (also called BSEP, bile salt export pump) located on chromosome 2q 24. PFIC3 is due to mutations of the MDR3 (multi drug resistance-3) gene encoding the biliary phospholipid transporter and is located on chromosome 7q 21(9). Though these new developments have made it possible to understand the underlying patho-physiology of cholestasis but they are not yet there for clinical diagnostic use or genetic counselling. Even in developed countries, despite intensive professional education in last 15-20 years, over 50% of children with neonatal cholestasis are referred late(4). Many of these children do not get benefit of the possible early intervention and progress to develop chronic liver disease leaving liver transplantation as the only option. With the advancements in the surgical technique, improved pre and post liver transplant management, and better understand-ing of immunosuppresive regimens, a 5-year survival of more than 90% has become a reality(10). We hope that the recommendations of the consensus report will improve the outcome of infants with cholestasis in India.
Sanjay Bansal,
1. Mieli-Vergani G, Howard ER, Portman B, Mowat AP. Late referral for biliary atresia–missed opportunities for effective surgery. Lancet 1989; I: 421-423. 2. Howard ER, Mowat AP. Extra hepatic biliary atresia. Recent developments in management. Arch Dis Child 1977; 52: 825-827. 3. Ohkohchi N, Chiba T, Endo N, Goto M, Ibrahim M. Long-term follow-up after surgery for patients with biliary atresia. J Pediatr Gastro-enterol Nutr 1989; 9: 416-420. 4. Mowat AP, Davidson LL, Dick MC. Earlier identification of biliary atresia and hepatobiliary disease: Selective screening in the third week of life. Arch Dis Child 1995; 72: 90-92. 5. Klebanoff HJ, Read JS, Mills JL, Shiono PH. The risk of childhood cancer after neonatal exposure to vitamin K. N Engl J Med 1993; 329: 905-908. 6. McClement JW, Howard ER, Mowat AP. Results of surgical treatment for extrahepatic biliary atresia in United Kingdom 1980-2. Br Med J 1985; 290: 345-347. 7. Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard J-L, Auvert B. Prognosis of biliary atresia in the era of liver trans-plantation: French National Study from 1986 to 1996. Hepatology 1999; 30: 606-611. 8. Pediatric Gastroenterology Subspecialty Chapter of Indian Academy of Pediatrics: Consensus Report on Neonatal Cholestasis Syndrome. Indian Pediatr 2000; 37: 845-851. 9. Jacquemin E, Hadchouel M. Genetic basis of progressive familial intrahepatic cholestasis. J Hepatology 1999; 31: 377-381. 10. Dhawan A, Muiesan P. Pediatric liver trans-plantation. Acta Pediatr Japonica 1999; 40: 525-529. |
![]()