|
|
|
Indian Pediatr 2018;55:
605-607 |
 |
A Unique Genomic Variant of HDR Syndrome in
Newborn
|
|
Ramesh Vidavalur 1,2
and Srisatish Devapatla1
From 1Cayuga Medical Center, Ithaca; and 2Department
of Clinical Pediatrics, Weill Cornell Medical College; New York, USA.
Correspondence to: Dr Ramesh Vidavalur, Division of
Neonatology, Department of Pediatrics, Cayuga Medical Center, 101 Dates
Drive, Ithaca NY 14850.
Email: rvidavalur@ yahoo.com
Received: February 08, 2017;
Initial Review: June 19, 2017;
Accepted: May 09, 2018.
|
Background: HDR syndrome (also known as Barakat
syndrome) is a rare genetic disorder due to deletions/mutations on
specific regions of zinc-finger transcription factor (GATA3)
gene. Case Characteristics: A male preterm infant presented with
multiple dysmorphic features characterized by small for gestational age,
hypognathia and facial abnormalities. Observation: Investigations
revealed hypocalcemia and low parathyroid hormone levels and bilateral
sensorineural deafness. Outcome: Chromosomal microarray analysis
revealed a combination of deletion on chromosome 10p (10p15.3p14) with
loss of GATA3 gene and duplication of chromosome 20p (20p13p12.3)
as a result of unbalanced 10:20 translocation. Message: Detecting
this syndrome at neonatal age is very important because it allows early
intervention to minimize future clinical problems.
Keywords: Barakat syndrome, Deafness, Hypoparathyroidism,
Renal dysplasia.
|
|
H
ypoparathyroidism, deafness, and renal dysplasia
syndrome (HDR syndrome also known as Barakat syndrome) is an autosomal
dominant disorder with symptoms of hypocalcemia and proteinuria [1].
Phenotypical features are attributed to mutations in the GATA3
gene on the short arm of chromosome 10, as GATA3 encodes a
transcription factor that is essential for embryonic development of the
parathyroid glands, auditory system and kidneys [2]; although, a wide
range of phenotypes have been described [3]. We report a unique case of
HDR syndrome in a preterm infant with a molecular diagnosis of
unbalanced 10;20 translocation involving a combination of loss of
function mutation of GATA3 on chromosome 10p and duplication of
chromosome 20p.
Case Report
A male, small for gestational age newborn with birth
weight of 1845g was delivered at 35 weeks gestation to a 21-year-old
second para mother by Caesarian section secondary to breech presentation
and category-2 fetal heart tracing. There was no family history of
developmental delay or inherited disorders. Parents were
non-consanguineous. Rupture of membranes was noted one hour prior to
delivery and meconium stained amniotic fluid was also observed. Infant
was delivered by breech extraction and there was a considerable
difficulty in delivering the head. He was pale, limp and apneic at
birth. He needed chest compressions, endotracheal intubation, and
positive pressure ventilation in delivery room. Apgar scores were 1,5
and 8 at 1-, 5- and 10- minutes, respectively.
Physical examination revealed hypotonia and
dysmorphic features characterized by wide nasal bridge, down slanting
eyes, high arched palate, micrognathia, low set ears, tapered fingers
and bilateral overlapping of toes (Fig. 1). He was
extubated to continuous positive airway pressure (CPAP). He required
CPAP for 10 hours and was treated with ampicillin and gentamicin for 48
hours. His tone improved after 48 hours of life. Full enteral feeds with
expressed breast milk were achieved by 4th day of life. A grade 2/6
late-systolic murmur was heard along left sternal border; echocardiogram
confirmed small muscular ventricular septal defect.
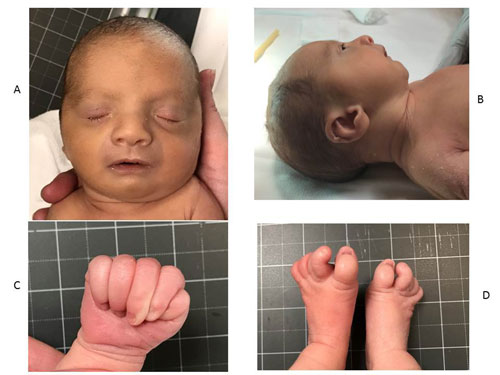 |
|
Fig. 1 (a) Frontal view showing down
slanting eyes, broad nasal bridge, prominent forehead; (b)
Lateral view showing low set, posteriorly rotated ears with
prominent pinna, anteverted nose and micrognathia; (c) Hand
showing tapered fingers; and (d) Bilateral feet showing
prominent 2 nd toe and overlapping
of 2nd and 3rd toe with sandal gap.
|
Initial complete blood count was within normal
limits. His metabolic panel was notable for hypocalcemia with serum
calcium of 7.2 mg/dL. His discharge calcium level was 8 mg/dL with
ionized calcium level of 4.17 mg/dL (Normal 5.1-5.9 mg/dL) and
phosphorous level of 9 mg/dL (Normal 4.3-5.4 mg/dL). His parathyroid
hormone level at 12 days of life was 13 pg/mL.
The chromosomal microarray analysis revealed a
pathogenic deletion from chromosome 10p, including ZMYND11 gene
(OMIM #608668) and GATA3 gene (OMIM #131320). The analysis also
revealed duplication of 20p chromosome. Fluorescent in situ
hybridization (FISH) studies using 10p and 20p subtelomeric probes
identified that these abnormalities were the result of an unbalanced 10;
20 translocation.
Bilateral sensorineural hearing loss was confirmed
with brainstem auditory evoked response testing. Infant was started on
low phosphorous formula after consultation with endocrinologist.
Discussion
Many cases of HDR syndrome have been described at
different ages and often diagnosed after the onset of symptoms such as
hypocalcemia and deafness [4], and associated renal anomalies [5].
Previous studies have shown that mutations or
terminal deletions of GATA3 gene on telomeric portion of 10p
chromosome (10p14) are responsible for characteristic phenotype of this
syndrome [6]. In addition to features of Barakat syndrome, large
deletions on 10p chromosome have also been associated with developmental
delay, intellectual disability, autistic behavior, heart defects and
immune deficiency [7,8]. The presence and significance of 20p
duplication in combination with subtelomeric deletion of chromosome
10p15.3 to p14 have not been reported in literature to the best of our
knowledge.
Detailed haplogenetic studies suggested terminal
deletion of chromosome 10p15 region involves ZMYND11 gene and
this deletion is associated with characteristic phenotype of high arched
palate, webbed toes, intrauterine growth restriction, cognitive
deficits, speech disorders, hypotonia and intellectual disability [8,9].
Interestingly, our infant showed normal kidneys with good function. This
may be due to low penetrance for renal anomalies as described in some of
the previous published studies. It is possible, therefore, that GATA3
mutations are associated with a relatively varied penetrance and
expressivity of the HDR triad features. Clinical features, including
hypertelorism, micrognathia and hypotonia have been earlier reported in
20p duplication [10].
The combined information of present and previous
published cases suggests that terminal deletion of chromosome 10p14-p15
represent a syndrome with a distinct severe phenotype than previously
described.
Acknowledgements: Nicole L Hoppman, Mayo Medical
Laboratory for molecular cytogenetics analysis and valuable inputs.
Contributors: RV: clinical care, carried out
literature search, drafted the initial and revised manuscript; SD:
clinical care, reviewed and revised the manuscript. Both authors
approved the final version of manuscript.
Funding: None; Competing Interest: None
stated.
References
1. Barakat AY, Dalbora JB, Martin MM, Jose PA.
Familial nephrosis, nerve deafness, and hypoparathyroidism. J Pediatr.
1977;91:61-4.
2. Esch HV, Groenen P, Nesbit MA, Schuffenhauer S,
Lichtner P, Vanderlinden G, et al. GATA3 haplo-insufficiency
causes human HDR syndrome. Nature. 2000;406:419-22.
3. Muroya K. GATA3 abnormalities and the
phenotypic spectrum of HDR syndrome. J Med Genet. 2001;38:374-80.
4. Hasegawa T, Hasegawa Y, Aso T, Koto S, Nagai T,
Tsuchiya Y, et al. HDR syndrome (hypoparathyroidism,
sensorineural deafness, renal dysplasia) associated with del(10)(p13).
Am J Med Genet. 1997;73:416-8.
5. Kato Y, Wada N, Numata A, Kakizaki H. Case of
hypoparathyroidism, deafness and renal dysplasia (HDR) syndrome
associated with nephrocalcinosis and distal renal tubular acidosis. Int
J Urol. 2007;14:440-2.
6. Lichtner P. An HDR (hypoparathyroidism, deafness,
renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome
region on 10p13/14. J Med Genet. 2000;37:33-7.
7. Lindstrand A, Malmgren H, Verri A, Benetti E,
Eriksson M, Nordgren A, et al. Molecular and clinical
characterization of patients with overlapping 10p deletions. Am J Med
Genet A. 2010;152A:1233-43.
8. Cobben J, Weiss M, Dijk FV, Reuver RD, Kruiff CD,
Pondaag W, et al. A de novo mutation in ZMYND11, a candidate gene
for 10p15.3 deletion syndrome, is associated with syndromic intellectual
disability. Eur J Med Genet. 2014;57:636-8.
9. Descipio C, Conlin L, Rosenfeld J, Tepperberg J,
Pasion R, Patel A, et al. Subtelomeric deletion of chromosome
10p15.3: Clinical findings and molecular cytogenetic characterization.
Am J Med Genet A. 2012;158A:2152-61.
10. Izumi K, Kubota N, Arakawa M, Takayama M, Harada
Y, Nakamura T, et al. Dissecting the phenotype of supernumerary
marker chromosome 20 in a patient with syndromic pierre robin sequence:
Combinatorial effect of gene dosage and uniparental disomy. Am J Med
Genet Part A. 2015;167:1289-93.
|
|
|
 |
|
